A Molecular Dynamic Approach to a Theory on the Dynamical Behaviour of HIV (3LPU Protein) - Water Molecules Interactions in Atomic Structures
1 State key laboratory of Fine Chemicals, Department of Pharmaceutical Science, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, People’s Republic of China
2 Department of Neurology, Kermanshah University of Medical Science, Kermanshah, Iran
3 Health Center of Tuyserkan, Hamadan University of Medical Science, Hamadan, Iran
4 School of Physics & School of Microelectronics, Dalian University of Technology, Dalian 116023, People’s Republic of China
5 Kermanshah University of Medical Science, Kermanshah, Iran
6 School of Energy and Power Engineering, Dalian University of Technology, Dalian, People's Republic of China
* Corresponding author. E-mail: bitafarhadi@mail.dlut.edu.cn
Received: Apr. 13, 2022; Accepted: Oct. 7, 2022; Published: Oct. 22, 2022
Citation: Zahra Karimi, Shokoufeh Heydaripour, Rozita Farhadi, Bita Farhadi, Fariba Karami Moghadam, and Mohammad Omidi, A Molecular Dynamic Approach to a Theory on the Dynamical Behaviour of HIV (3LPU Protein) - Water Molecules Interactions in Atomic Structures. Nano Biomed. Eng., 2022, 14(2): 159-166.
DOI: 10.5101/nbe.v14i2.p159-166
Abstract
Molecular dynamics simulation was used to investigate the structure of the H2O molecule and its dynamical behaviour near an HIV (3LPU protein). This study simulated the atomic interaction between 3LPU protein and H2O molecules using a precise atomic arrangement. The interaction between 3LPU protein and H2O molecules is influenced by temperature and pressure. According to our simulated findings, the amplitude of atomic oscillation increases as the atom's temperature rises to 400 K. As a result of this occurrence, the interatomic force of structures increases. As a result, as the temperature rises, the diffusion coefficient of H2O molecules into 3LPU protein changes from 0.421 to 0.861 μm2/ms. The dynamical behaviour of atomic structures is also influenced by pressure. The diffusion coefficient of H2O molecules into the 3LPU protein structure fell from 0.587 to 0.052 μm2/ms when the pressure of the simulated structures was increased from 0 to 4 bar.
Keywords: Molecular dynamics, 3LPU protein, temperature, pressure, dynamical behaviour.
Introduction
AIDS/HIV infection is a worldwide epidemic, with instances documented in almost every nation. According to the Joint United Nations Program on HIV/AIDS, an estimated 36.7 million people were living with HIV by the end of 2016 [1, 2]. 95% of HIV/AIDS patients live in low- and middle-income countries; 50% are women, and 2.1 million are children under the age of 15. The distribution of these instances throughout the country. The incidence of HIV infection has grown more than fourfold since 1990, owing to the ongoing high rates of new HIV infections and the life-prolonging benefits of antiretroviral medication. The worldwide prevalence of HIV infection among persons aged 15 to 49 years was 0.8% in 2016, with rates ranging significantly by nation and location[3-5].
In 2016, an estimated 1.8 million new cases of HIV infection were reported globally, with 160,000 of those infections occurring in children under the age of 15 years; around one-third of new infections occurred in persons aged 15 to 24 years. Heterosexual transmission is responsible for the majority of new HIV infections worldwide. Those who belong to particular high-risk groups are disproportionately impacted[6, 7].
Globally, the expected yearly number of new HIV infections declined by 40% between 2000 and 2016. These decreases in worldwide HIV incidence are most likely due to success in HIV prevention efforts and increasing antiretroviral medication availability to HIV-infected persons, which renders them far less likely to transfer the virus to sexual partners. Between 2010 and 2016, the projected adult incidence decreased by 11%. Between 2010 and 2016, there was a -47% decrease in HIV infections among children aged 15 years, owing partly to the increased availability of antiretroviral drugs to prevent HIV transmission from mother to child[8, 9].
Human and animal urine and feces contain pathogenic microorganisms that pollute water and cause water-borne illnesses. Infections may develop in those who drink this polluted water[10]. Viruses may harm every living entity, including plants, microbes, animals, and humans. Viruses may interact with their hosts in a variety of ways, depending on whether the virus is host-specific (HIV) or not (influenza)[11]. Viruses may spread via various methods; for example, influenza is airborne and spreads by inhalation of infected air, but HIV is spread directly through contaminated bodily fluids. Enteric viruses are spread by ingesting infected food or water contaminated with feces, and they mostly damage the intestines. HIV infection caused a variety of illnesses, including HIV infections and acquired immune deficiency syndrome (HIV/AIDS)[12, 13]. After being infected, the patient may have no symptoms or a brief period of influenza-like sickness[14]. The patient will be asymptomatic for a long time in the following stage[15]. As the illness develops, the immune system becomes more weakened, increasing the chance of contracting common diseases such as TB and other opportunistic infections[16]. Concerning AIDS therapy is one of the most difficult tasks in medicine. As a result, research into how HIV interacts with the aquatic environment in which it is transmitted at varying temperatures and pressures is required. Figure 1 shows the structure of a key component of this virus (3LPU protein)[17].
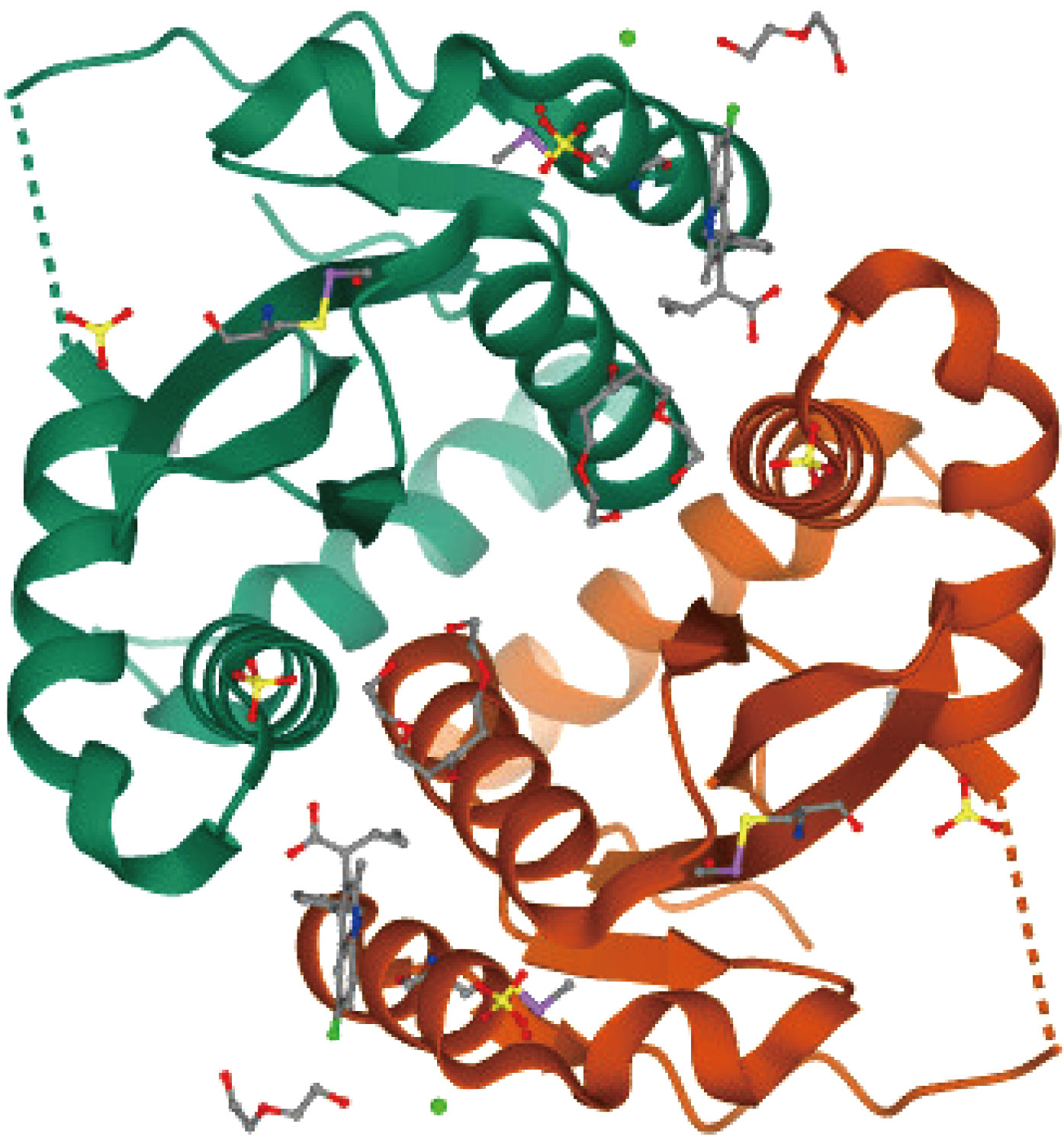
Fig. 1 Schematic of a crucial component of the HIV (3LPU protein) structure.
The 3LPU protein is one of the most critical components of HIV. As a result, understanding its atomic structure is crucial. The atomic structure of the 3LPU chain was revealed by Ding et al. [18]. As a result, we focused on this aspect of HIV's interaction with H2O molecules in our study. Furthermore, Molecular Dynamics (MD) simulation studies modeling particle movements in biological fields and causing fluctuations in the relative locations of atoms in proteins, viruses, and other atomic structures as a function of time[19-21]. Good results may be obtained even when the MD strategy is combined with other approaches [22]. In previous works, Muhammad Ibrahim [23] Molecular dynamics simulation was used to investigate the dynamical behaviour of 3LPT protein-water molecule interactions in atomic structures. In another work, Jun and Zhao [24] The quantum mechanical/molecular mechanical (QM/ MM) technique was used to explore two occurrences of monohydrate and polyhydration of H2O molecules. The obtained binding energy between the water molecules is smaller than the hydration energy, according to the simulation findings. Further, Yin and Zhao [25] Look into the polar head group of the dipalmitoylphosphatidylcholine (DPPC) molecule in both the gas phase and aqueous solution. They demonstrated that the hybrid QM/MM approach is an accurate way of describing the conformations of the DPPC headgroup[26].
A technical grasp of atom trajectories allows us to see biological phenomena such as molecules, atomic structures, and interactions between various structures. In the late 1950s, Monte Carlo's success in simulations led to the creation of MD[27-29]. Laplace's fundamental work laid the groundwork for the development of MD in the past[30]. Alder and Wainwright later presented MD simulations as a useful tool for studying atomic structures [21].
MD modelling is now widely utilised in biology and biophysics to investigate atomic structures [31-33].
Temperature changes are essential for bio structures because different temperatures affect bio structures during the day, and also, there are different temperatures in different environments. Changes in pressure because the pressure is additional at different heights and biostructures show other behaviors at different pressures; it is essential to investigate the effect of pressure on biostructures. Theoretical studies to anticipate HIV (3LPU protein) and H2O molecules interactions in different temperatures (300 K – 400 K) and pressures (0–4 bar) were done in this research using MD simulation for first time. Physical factors such as total energy, diffusion coefficient, and volume will be presented to forecast atomic structure behavior.
Ibrahim et al. [22] reported temperature increasing causes changing the diffusion coefficient from 0.74 to 0.91 μm2/s in H2O molecules into 3LPT protein. Pressure also has a significant effect on the dynamical behavior of atomic structures. In this work, as the temperature rises from 300 to 400 K, the diffusion coefficient of H2O molecules in the 3LPU protein/ H2O combination increases from 0.42 to 0.86 μm2/s. Our results are closed to the data that has already been reported.
Computational method
The atomic interaction between 3LPU protein and H2O molecules was simulated using the MD technique at a temperature range of 300 K to 400 K and a pressure range of 0–4 bar. Furthermore, the activation volume is 4641 Å3. The attraction force between distinct sections of the new structure falls drastically when the volume of the atomic structure is increased beyond this threshold. Hence, physical stability is reduced to a minimal rate. MD simulations are one of the most important tools in the theoretical study of biological molecules. Calculating the time-dependent behaviour of a chemical system via this computational approach provides detailed information on changes in atomic structures[27]. These approaches are now routinely used to investigate biological molecules' structure, dynamics, and thermodynamics and their complexes. Particles interact with one another utilising suitable atomic and interatomic force-field (IFF) models. Structures and the quantity of IFF the atoms of HIV have been modelled in this work utilising Dreiding and UFF (Universal Force Field)[34, 35]. The dynamics of biological, organic, and the major group inorganic have also been predicted using Dreiding and UFF force fields. Furthermore, the Leonard-Jones potential (LJ) has been used to analyse atoms' non-bond interactions[36, 37]:
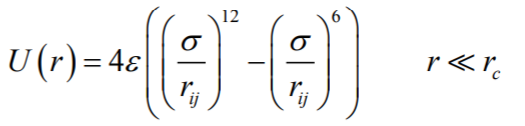 (1)
(1)
where ε represents the depth of the potential well, σ shows the finite distance at which the interatomic potential is zero, and the distance between the atoms is represented by r. In this simulation, the cut-off radiusis considered to be rc = 12 Å. The σ and ε values obtained are represented in Table 1 where ε denotes the potential well's depth, σ denotes the finite distance at which the interatomic potential is zero, and r is the distance between the atoms. The cut-off radius is assumed to be rc = 12 Å in this simulation. Table 1 shows the ε and σ values that were acquired.
Table 1 In current MD simulations, the ε and σ constants in the LJ potential.
Atom | σ (Å) | ε (kcal/mol) |
As | 4.230 | 0.309 |
C | 4.180 | 0.305 |
Ca | 3.42 | 0.050 |
Cl | 3.914 | 0.305 |
H | 3.200 | 0.010 |
N | 4.000 | 0.415 |
O | 3.600 | 0.415 |
S | 4.140 | 0.215 |
The bond strength and bond-angle bend parameters make up the bonded interactions. The bond strength is characterised in the current model by a simple harmonic oscillator using the following equation[37]:
![]() (2)
(2)
The harmonic oscillator constant kr and the atomic bond length r0 represent the harmonic oscillator constant and the atomic bond length, respectively. Table 2 shows the bonded interactions of the harmonic oscillator constant and atomic bond lengths. Furthermore, using a simple angular harmonic oscillator, the following equation represents the bond-angle bend in 3LPU protein[38]:
![]() (3)
(3)
kθ and θ0 denote the angular harmonic oscillator constant and the angle's equilibrium value, respectively. Equilibrium distance, harmonic constants.
Table 2 In MD simulation, the harmonic constants and equilibrium distance of different bindings were employed[33, 34].
Bond | Kr (Kcal/mol) | r0 (Å) |
As-C | 700 | 1.970 |
As-O | 700 | 1.860 |
C-C | 700 | 1.530 |
C-Cl | 700 | 1.757 |
C-N | 700 | 1.462 |
C-O | 700 | 1.420 |
C-S | 700 | 1.800 |
H-O | 700 | 0.980 |
N-N | 700 | 1.394 |
N-O | 700 | 1.352 |
O-O | 700 | 1.310 |
O-S | 700 | 1.690 |
In our MD simulations, we used the Simple Point Charge (SPC) model to represent H2O molecules [39]. Three locations in this model represented the electrostatic interactions. A negative charge was added on the oxygen atom to balance the partial positive charges on the hydrogen atoms.
The atomic bond length of OH and the equilibrium angle of H-O-H in this model are 1.0 Å and 109.47°, respectively. Furthermore, all hydrogen atom interatomic interactions are disregarded.
The MD simulation technique was applied at the beginning condition after atomic modelling of each structure. Table 3 shows the angular harmonic constant and equilibrium degree for atomic structures in the 3LPU protein.
Table 3 In the simulation box, the angular harmonic constant and equilibrium degree for atomic structures [33, 34].
Angle | Kθ (Kcal/mol) | θ0 |
C-As-O | 100 | 92.100 |
C-C-C | 100 | 109.471 |
C-C-Cl | 100 | 109.471 |
C-C-N | 100 | 109.471 |
C-C-O | 100 | 109.471 |
C-C-S | 100 | 109.471 |
C-N-C | 100 | 106.700 |
C-N-N | 100 | 106.700 |
C-N-O | 100 | 106.700 |
C-O-C | 100 | 104.510 |
C-O-N | 100 | 104.510 |
To determine the dynamical behaviour of particles, Newton's equation is numerically solved by the following equation. [23]:
![]() (4)
(4)
![]() (5)
(5)
Fi is the total force, mi is the atomic mass, ri is the position, and vi is the velocity of atom I in this equation. The forces between particles are computed in MD simulations using interatomic potential. Technically, we used the LAMMPS software to do MD simulations for the 3LPU protein and H2O molecules in our research[40-43]. LAMMPS is a molecular dynamics simulator that specialises in material modelling. In this research, MD simulation was carried out using the LAMMPS program in two steps:
Step 1: HIV 100 Å length in x, y, and z-directions and periodic boundary conditions enforced in all of these directions in the following MD computations[31, 35].
The temperature of the atomic structures was equilibrated at 300, 350, 375, and 400 K with a 1 fs time step in the following MD simulations. The Nose-Hoover thermostat with 0.01 damping rate (Δt = 0.01) for 1 ns HIV was utilised in the temperature equilibration procedure[36, 39, 40]. The total energy of simulated structures was given in this stage to ensure that the atomic model and interatomic force field were correct. The computational running was then maintained for 1 ns after the system attained equilibrium temperature and overall energy rate.
Step 2: The simulated 3LPU protein structure was equilibrated for 1 ns at 300, 350, 375, and 400 K in the second step. Physical metrics such as total energy, structure distance, diffusion coefficient, and volume of HIV structures were published to analyse the atomic interaction between these structures.
The conjugate gradient (CG) method was used to optimize the potential energy of the structure. For this simulation, the real units, full atomic style, periodic boundary, and Newton’s method were used. The structure of the 3LPU protein and H2O molecules are simulated in the middle of the box. The simulated box is filled with 30,000 H2O molecules, and 3LPU protein includes 1340 molecules. This simulation is done in a three-dimensional box with x, y, and z = 200 Å dimensions.
Results and discussion
Equilibration of the atomic structure of the 3LPU protein
The atomic structure of isolated 3LPU protein was researched in the first portion of this study, and the dynamical behaviour of 3LPU protein and H2O molecules was explored in the latter section. Figure 2 depicts the atomic configuration of the 3LPU protein, which contains the elements As, Ca, Cl, C, O, N, and S. To save time in our MD simulations, we avoided repeating the 3LPU protein and instead investigated one full atomic unit of this virus. By adding periodic boundary constraints in our MD simulations, we anticipated our findings to be comparable to the real size of the 3LPU protein.
Geometry minimization of atomic structure was done using the conjugate gradient approach before the equilibration phase, as previously described. In big sparse systems, this strategy is often utilised.
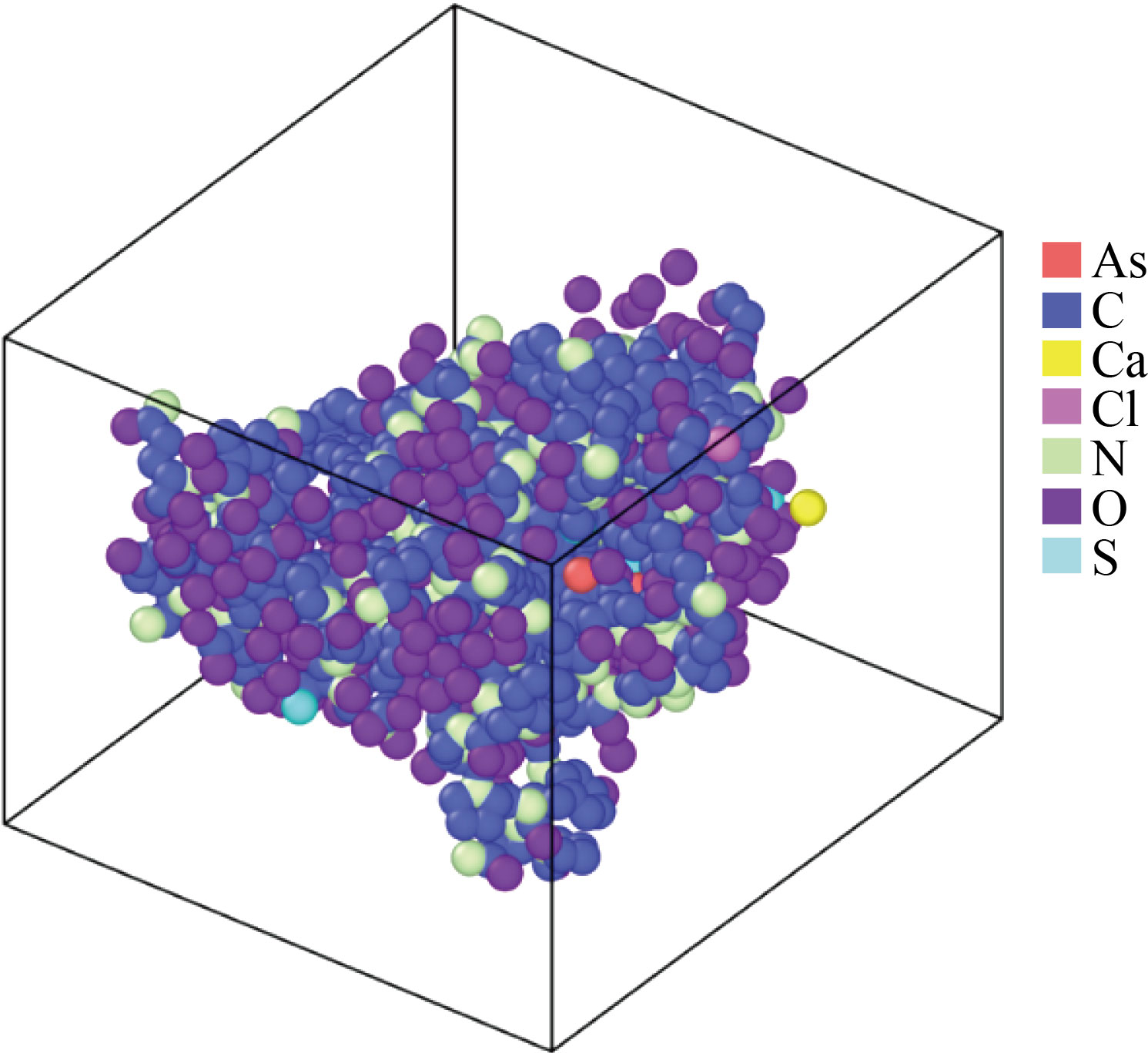
Fig. 2 In the NVE ensemble, a simulated atomic representation of the 3LPU protein.
The total energy estimated for 3LPU protein demonstrates that it is stable at 300, 350, 375, and 400 K. Figure 3 depicts the variations in total energy of the 3LPU protein as a function of temperature and pressure.
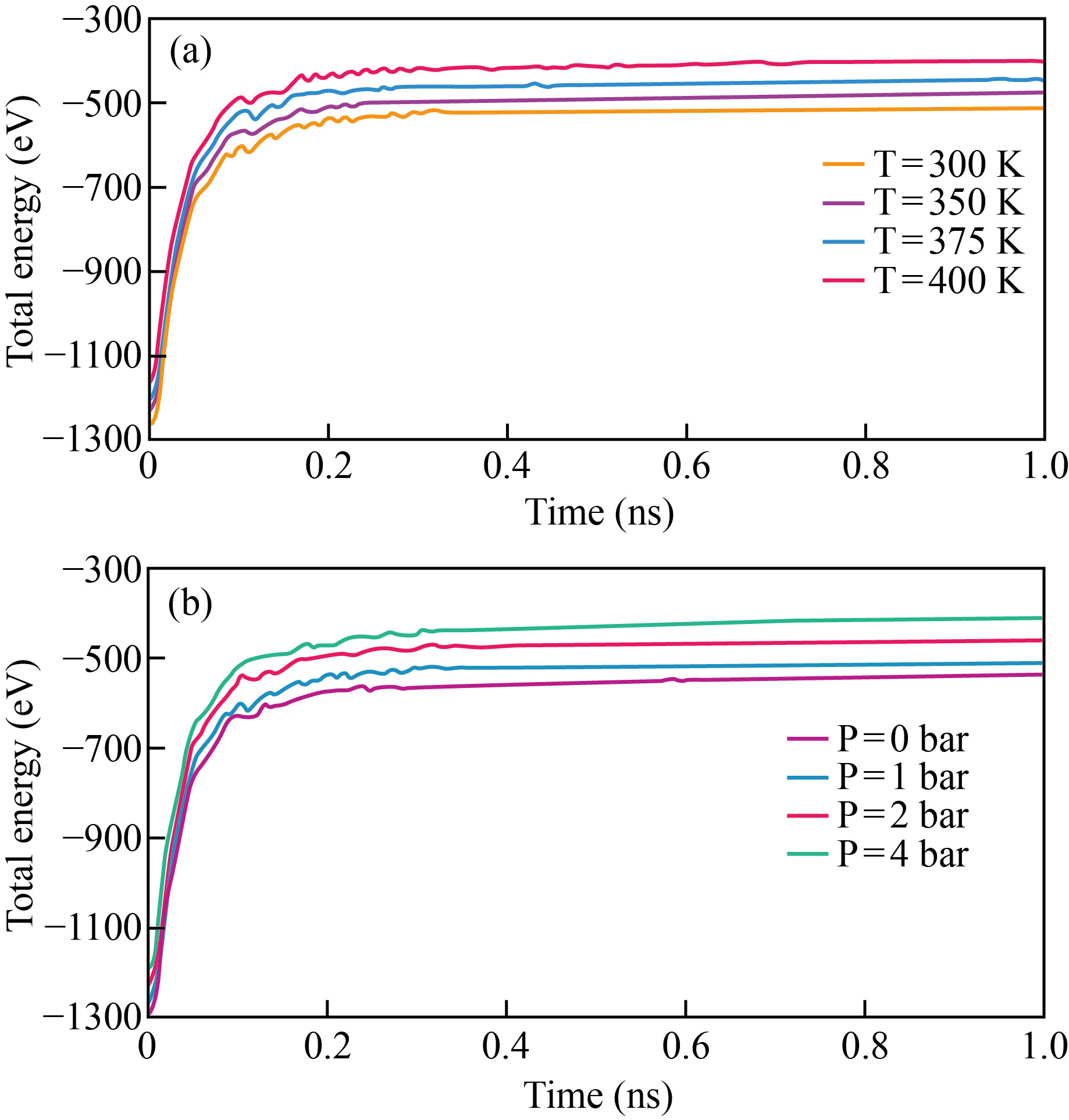
Fig. 3 3LPU protein total energy fluctuation as a function of a) temperature and b) pressure.
When the temperature is increased from 300 K to 400 K, the total energy decreases from -510.54 to -398.08 eV. The decrease in total energy of 3LPU protein caused by increasing temperature is due to increased atom mobility. As the mobility of the atom increases, the distance between atoms increases, and the total/potential energy of viral atoms drops. We may claim that when the temperature of the 3LPU protein rises, the structure's stability diminishes. Similar to temperature calculation, Fig. 3b depicts total energy convergence after 1 ns simulation. As pressure rises, it has an impact on the overall energy value. By raising pressure from 0 to 4 bar, the total energy value of 3LPU protein changes from -534.08 to -409.259 eV.
3LPU protein/H2O structure equilibration
The NVE ensemble was used to model the atomic interaction between 3LPU protein and H2O molecules in the following portion of our MD simulations. After modelling the 3LPU protein and H2O molecules, the MD simulation was conducted for 1 ns to equilibrate the thermodynamics. The structure of the 3LPU protein and H2O molecules are simulated in the center of the box, as illustrated in Fig. 4.

Fig. 4 Atomic depiction of a 3LPU protein/H2O combination that has been simulated.
30,000 H2O molecules are strewn over the simulated enclosure. OVITO program visualises these earliest atomic formations. At various temperatures and pressures, the total energy of the 3LPU protein/H2O combination fluctuates (Fig. 5). The negative value of atomic structures shows the mixture stability at simulated temperatures and pressures. Temperature and total energy are inversely related, with rising temperature resulting in a simulated atomic portrayal of a 3LPU protein/H2O combination, in the 3LPU protein/H2O mixture's total energy. Furthermore, raising the pressure from 0 to 4 bar reduces the total energy of the combination from -2554.11 eV to -2070.52 eV, lowering the stability of 3LPU protein /H2O.
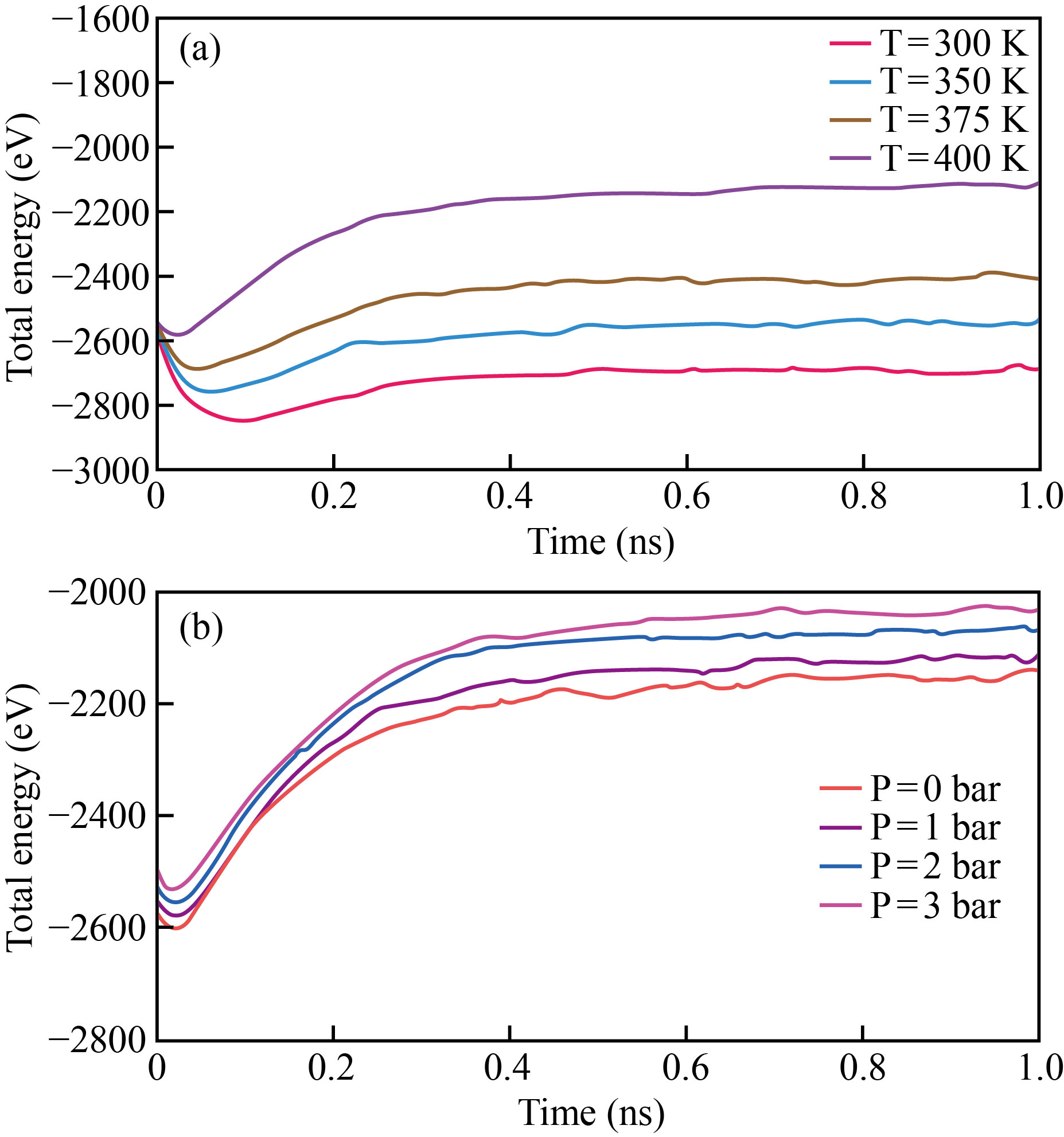
Fig. 5 The fluctuation in total energy of a 3LPU protein/H2O combination as a function of a) temperature and b) pressure.
Interaction of the 3LPU protein with H2O molecules on an atomic level
The initial mean squared displacement of the 3LPU protein /H2O combination was computed using the LAMMPS software for diffusion coefficient computation. Finally, the MSD parameter slope equals the H2O molecule transport coefficient to the 3LPU protein. Table 4,5 shows how the solvent's diffusion coefficients vary as temperature and pressure increase. The diffusion coefficient rises as temperature and pressure increase, according to these MD simulation data.
Table 4 Temperature-dependent diffusion coefficient of the solvent in simulated structures.
Temperature (K) | Diffusion coefficient (µm2/s) |
300 | 0.42 |
350 | 0.62 |
375 | 0.73 |
400 | 0.86 |
The findings reveal that raising the temperature from 300 to 400 K increases the amplitude of atomic oscillations. As a consequence, the diffusion coefficient of H2O molecules rises from 0.42 to 0.86 μm2/s in the simulated protein. As previously stated, raising the pressure has a similar effect. The diffusion coefficient reduces from 0.58 to 0.05 μm2/s when pressure rises from 0 to 4 bar (Table 5).
Table 5 The solvent diffusion coefficient of simulated structures as a function of pressure.
Pressure (bar) | Diffusion coefficient (µm2/s) |
0 | 0.58 |
1 | 0.42 |
2 | 0.25 |
4 | 0.05 |
Finally, as a function of MD simulation time steps, rising temperature and pressure trigger atomic destruction of the 3LPU protein. The last parameter investigated in our MD simulations is the volume changes of 3LPU protein after interacting with H2O molecules. We may deduce from Fig. 6 that the volume of 3LPU protein grows as time passes. The volume of the virus changes from 5485 Å3 to 6056 Å3 when the temperature of the 3LPU protein/H2O combination is raised. As the volume of the 3LPU protein increases, the atomic stability of the structure diminishes.
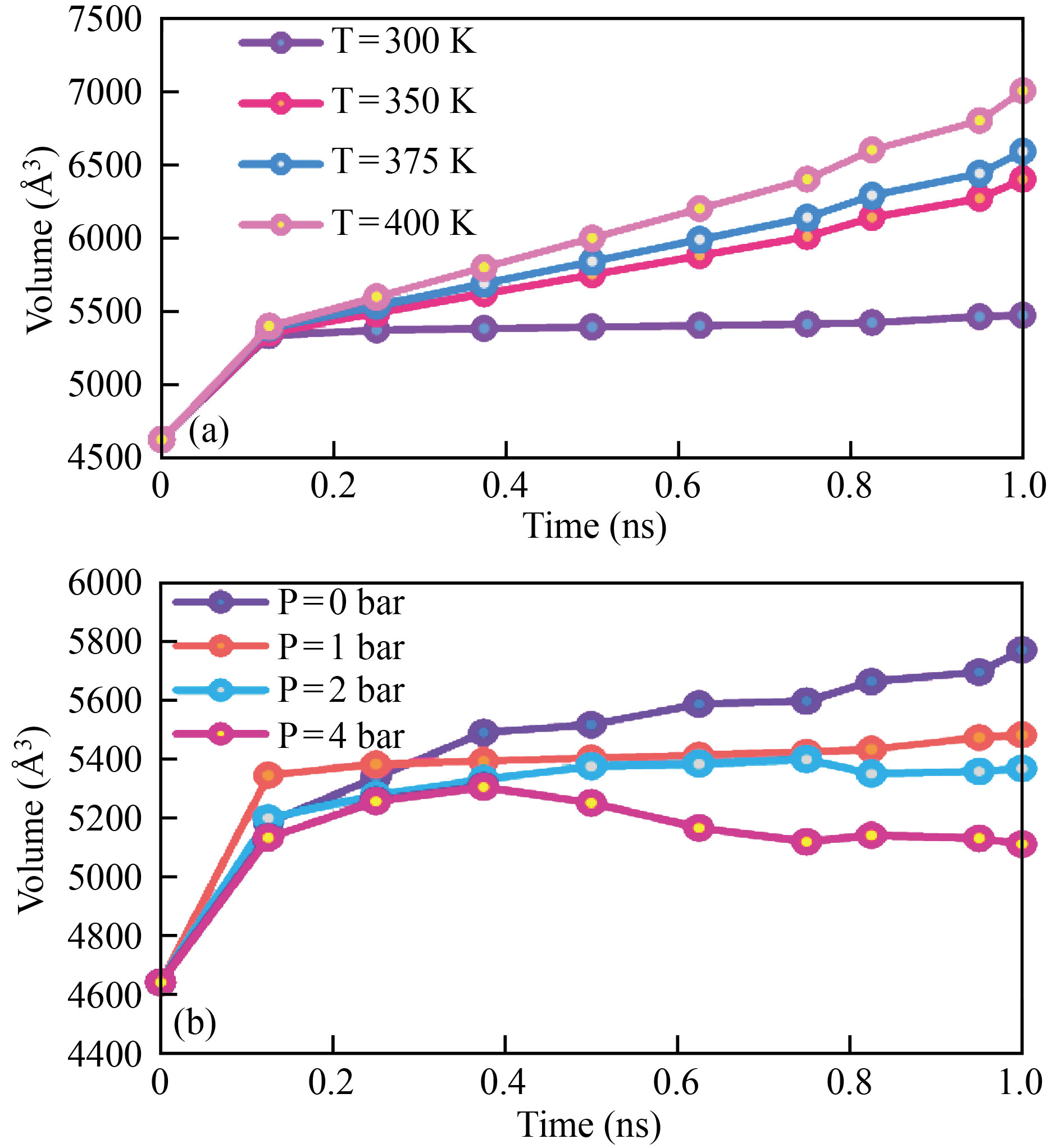
Fig. 6 The volume of 3LPU protein changes over time as a function of a) temperature and b) pressure.
By raising the volume of 3LPU protein (by increasing the temperature), the interatomic distance of this structure grows as a function of time, lowering the potential/total energy of 3LPU protein and so destroying this virus (see Fig. 6a). The volume of the atomic structure of 3LPU protein changes from 5667 Å3 to 5143 Å3 when the pressure is increased from 0 to 4 bar (Fig. 6b).
The amplitude of atomic oscillations increases as temperature and pressure factors increase, resulting in this atomic behaviour.
Conclusion
The impact of temperature and pressure variations on the interaction of 3LPU protein with H2O molecules was examined in this work. The precise atomic configuration of 3LPU protein and H2O molecules was simulated in our simulations. Interatomic interactions of the 3LPU protein / H2O combination are also described using UFF and Dreiding force fields. Finally, the following are the computational outputs from this simulation:
A. Using UFF and Dreiding force fields as an appropriate interatomic potential of the 3LPU protein / H2O mixture structure is efficient in this simulation.
B. As the temperature rises from 300 to 400 K, the diffusion coefficient of H2O molecules in the 3LPU protein/ H2O combination increases from 0.42 to 0.86 μm2/s.
C. As pressure is increased from 0 to 4 bar, the diffusion coefficient of H2O molecules in the 3LPU protein/ H2O combination rises from 0.58 to 0.05 μm2/s.
D. As H2O molecules diffuse into this atomic structure, the volume of 3LPU protein grows, and as this parameter rises, the atomic stability of 3LPU protein diminishes.
Conflict of Interests
The authors declare that no competing interest exists.
References:
1.HIV/AIDS., J.U.N.P.o. and W.H. Organization, 2008 report on the global AIDS epidemic. 2008: World Health Organization.
2.Latha, G.M. and E.S.N. Murthy, Clinical study on ocular manifestations in HIV/AIDS relation to CD4 count in tertiary care hospital, Andhra Pradesh. virus. 11: p. 12.
3.Nguyen, P.T., et al., Factors associated with high-risk behaviors of people newly diagnosed with HIV/AIDS: results from a cross-sectional study in Vietnam. AIDS care, 2021. 33(5): p. 607-615.
4.Diehr, M.C., et al., The 50 and 100-item short forms of the Paced Auditory Serial Addition Task (PASAT): demographically corrected norms and comparisons with the full PASAT in normal and clinical samples. Journal of clinical and experimental neuropsychology, 2003. 25(4): p. 571-585.
5.HIV/AIDS., J.U.N.P.o. and W.H. Organization, AIDS epidemic update, December 2006. 2006: World Health Organization.
6.MacNeil, A., et al., Global epidemiology of tuberculosis and progress toward achieving global targets—2017. Morbidity and Mortality Weekly Report, 2019. 68(11): p. 263.
7.Corbett, E.L., et al., The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Archives of internal medicine, 2003. 163(9): p. 1009-1021.
8.Organization, W.H., Report on global sexually transmitted infection surveillance 2018. 2018.
9.Koppaka, R., Ten great public health achievements--worldwide, 2001-2010. 2011.
10.Edition, F., Guidelines for drinking-water quality. WHO chronicle, 2011. 38(4): p. 104-108.
11.Koonin, E.V., T.G. Senkevich, and V.V. Dolja, The ancient Virus World and evolution of cells. Biology direct, 2006. 1(1): p. 1-27.
12.Krämer, A., M. Kretzschmar, and K. Krickeberg, Modern infectious disease epidemiology: Concepts, methods, mathematical models, and public health. 2010: Springer.
13.Kirch, W., Encyclopedia of Public Health: Volume 1: A-H Volume 2: I-Z. 2008: Springer Science & Business Media.
14.Parkhurst, J.O., The Ugandan success story? Evidence and claims of HIV-1 prevention. The Lancet, 2002. 360(9326): p. 78-80.
15.Lyles, C.M., et al., Evidence–based HIV behavioral prevention from the perspective of the CDC's HIV/AIDS Prevention Research Synthesis Team. AIDS Education & Prevention, 2006. 18(supp): p. 21-31.
16.Rahier, J.-F., et al., European evidence-based Consensus on the prevention, diagnosis and management of opportunistic infections in inflammatory bowel disease. Journal of Crohn's and Colitis, 2009. 3(2): p. 47-91.
17.Mishra, S.K., et al., Role of the ubiquitin-like protein Hub1 in splice-site usage and alternative splicing. Nature, 2011. 474(7350): p. 173-178.
18.Christ, F., et al., Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nature chemical biology, 2010. 6(6): p. 442-448.
19.Karplus, M. and G.A. Petsko, Molecular dynamics simulations in biology. Nature, 1990. 347(6294): p. 631-639.
20.Liu, X. and Y.-P. Zhao, Generating artificial homologous proteins according to the representative family character in molecular mechanics properties–an attempt in validating an underlying rule of protein evolution. FEBS letters, 2010. 584(5): p. 1059-1065.
21.Jolfaei, N.A., et al., Investigation of thermal properties of DNA structure with precise atomic arrangement via equilibrium and non-equilibrium molecular dynamics approaches. Computer methods and programs in biomedicine, 2020. 185: p. 105169.
22.Alipour, P., et al., Molecular dynamics simulation of fluid flow passing through a nanochannel: Effects of geometric shape of roughnesses. Journal of Molecular Liquids, 2019. 275: p. 192-203.
23.Ibrahim, M., et al., Investigation of dynamical behavior of 3LPT protein-water molecules interactions in atomic structures using molecular dynamics simulation. Journal of Molecular Liquids, 2021. 329: p. 115615.
24.Yin, J. and Y.-P. Zhao, Hybrid QM/MM simulation of the hydration phenomena of dipalmitoylphosphatidylcholine headgroup. Journal of colloid and interface science, 2009. 329(2): p. 410-415.
25.Yin, J. and Y.-P. Zhao, Molecular dynamics simulation of adhesion forces in a dipalmitoylphosphatidylcholine membrane. Surface Review and Letters, 2007. 14(04): p. 671-675.
26.Lemkul, J.A., et al., An empirical polarizable force field based on the classical drude oscillator model: development history and recent applications. Chemical reviews, 2016. 116(9): p. 4983-5013.
27.Fermi, E., J. Pasta, and S. Ulam, Los alamos report la-1940. Fermi, Collected Papers, 1955. 2: p. 977-988.
28.Alder, B.J. and T.E. Wainwright, Studies in molecular dynamics. I. General method. The Journal of Chemical Physics, 1959. 31(2): p. 459-466.
29.Rahman, A., Correlations in the motion of atoms in liquid argon. Physical review, 1964. 136(2A): p. A405.
30.Schlick, T., Pursuing Laplace’s vision on modern computers, in Mathematical Approaches to Biomolecular Structure and Dynamics. 1996, Springer. p. 219-247.
31.Ashkezari, A.Z., et al., Calculation of the thermal conductivity of human serum albumin (HSA) with equilibrium/non-equilibrium molecular dynamics approaches. Computer methods and programs in biomedicine, 2020. 188: p. 105256.
32.Tohidi, M. and D. Toghraie, The effect of geometrical parameters, roughness and the number of nanoparticles on the self-diffusion coefficient in Couette flow in a nanochannel by using of molecular dynamics simulation. Physica B: Condensed Matter, 2017. 518: p. 20-32.
33.Rappé, A.K., et al., UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American chemical society, 1992. 114(25): p. 10024-10035.
34.Mayo, S.L., B.D. Olafson, and W.A. Goddard, DREIDING: a generic force field for molecular simulations. Journal of Physical chemistry, 1990. 94(26): p. 8897-8909.
35.Lennard-Jones, J.E., The determination of molecular orbitals. Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences, 1949. 198(1052): p. 1-13.
36.Berendsen, H., J. Grigera, and T. Straatsma, The missing term in effective pair potentials. Journal of Physical Chemistry, 1987. 91(24): p. 6269-6271.
37.Farhadi, B., et al., Carbon doped lead-free perovskite with superior mechanical and thermal stability. Molecular Physics, 2021: p. e2013555.
38.Farhadi, B., et al., Influence of contact electrode and light power on the efficiency of tandem perovskite solar cell: Numerical simulation. Solar Energy, 2021. 226: p. 161-172.
39.Wang, L.-P., T.J. Martinez, and V.S. Pande, Supporting Information for: Building Force Fields-an Automatic, Systematic and Reproducible Approach.
40.Plimpton, S., Fast parallel algorithms for short-range molecular dynamics. Journal of computational physics, 1995. 117(1): p. 1-19.
41.Brown, W.M., et al., Implementing molecular dynamics on hybrid high performance computers–short range forces. Computer Physics Communications, 2011. 182(4): p. 898-911.
42.Brown, W.M., et al., Implementing molecular dynamics on hybrid high performance computers–Particle–particle particle-mesh. Computer Physics Communications, 2012. 183(3): p. 449-459.
43.Zheng, Y., et al., Potential energy and atomic stability of H2O/CuO nanoparticles flow and heat transfer in non-ideal microchannel via molecular dynamic approach: the green–Kubo method. Journal of Thermal Analysis and Calorimetry, 2021. 144(6): p. 2515-2523.
Copyright© Zahra Karimi, Shokoufeh Heydaripour, Rozita Farhadi, Bita Farhadi, Fariba Karami Moghadam, and Mohammad Omidi. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.