Evaluation of the Interaction of Curcumin and Nigella Sativa on Brain Antitumor Molecule Using an Equilibrium Dynamics Simulation Tool for Biomedical Applications
Nakisa Ghamari1, Shokoufeh Heydaripour2, Zahra Karimi3, Rozita Farhadi4*, Nazanin Ghamari5
1Department of Biology, Faculty of Science, Razi University, Kermanshah, Iran
2Department of Neurology, Kermanshah University of Medical Science, Kermanshah, Iran
3Department of Neurology, Kermanshah University of Medical Science, Kermanshah, Iran
4Health Center of Tuyserkan, Hamadan University of Medical Science, Hamadan, Iran
5School of Medicine, Kermanshah University of Medical Sciences, Kermanshah, Iran
*Corresponding author. E-mail: r.farhadi88@yahoo.com
Received: Jun. 07, 2022; Revised: Nov. 22, 2022; Accepted: Dec. 22, 2022; Published: Dec. 31, 2022
Citation: N. Ghamari, S. Heydaripour, Z. Karimi, et al. Evaluation of the interaction of curcumin and nigella sativa on brain antitumor molecule using an equilibrium dynamics simulation tool for biomedical applications. Nano Biomedicine and Engineering, 2022, 14(4): 349–359.
DOI: 10.5101/nbe.v14i4.p349-359
Abstract
Curcumin and nigellin-1.1's atomic interactions on brain antitumor molecule are significant in medical research. For the first time, Molecular dynamic (MD) simulations based on Newton's law were utilized to predict the destruction of brain antitumor structure by curcumin and nigellin-1.1 with structure in the current research. To depict the atomic development of curcumin, nigellin-1.1, and brain antitumor molecule, DREIDING and universal force fields are used to model C, H, N, O, and S atoms. We calculate the total energy, center of mass distance, diffusion coefficient, and volume of atomic structures to explain the atomic interaction between these structures. The calculated rates for these physical parameters reveal an attraction force between curcumin and brain antitumor structure, as well as nigellin-1.1 and brain antitumor structure, with COM distances between curcumin and brain antitumor structures varying from 1.16 Å to 1.14 Å after 2 ns, and COM distances between nigellin-1.1 structures varying from 2.01 Å to 1.93 Å after 2 ns. The volume of a brain antitumor increases structurally from 1.33×106 Å3 to 2.24×106 Å3 following atomic contact with curcumin, and increases structurally from 1.33×106 Å3 to 2.83×106 Å3 after atomic interaction with nigellin-1.1, indicating tumor eradication.
Keywords: Curcumin; Nigellin-1.1; Brain tumor; Molecular dynamic; Atomic interactions
Introduction
Lack of efficacy and adverse side effects are two important problems in the current therapeutic field, along with complicated cellular and molecular processes of diseases. Therefore, developing tools in computational methods that discover new formulations with fewer side effects and optimal doses is promising, to reduce the attrition rate and improve the drug discovery process [1]. Today, the use of various medicinal herbs has received a lot of attention in treating diseases such as cancer, so brain tumors are no exception [2]. Even with a long time of research, brain tumors continue to be many of the deadliest of all types of cancer. Primary brain tumors are a heterogeneous group of malignancies that originate in cells of the central nervous system. A sort of models tractable for preclinical research were evolved to recapitulate human brain tumors, permitting us to recognize the underlying pathology and discover capable treatments [3]. For instance, drosophila Melanogaster is a strong and effective model organism for surveying molecular and cellular mechanisms that underlie neurodegeneration, and plenty of genes and pathways with roles in neuroprotective and neurodegeneration had been recognized and characterized on this organism [4]. The drosophila TRIM-NHL protein brain tumor (Brat) intuits differentiation of neuronal stem cells by suppressing self-renewal factors. Brat is an RNA-binding protein and functions as a translational repressor and acts as a growth suppressor in the larval brain. TRIM-NHL proteins are protected between metazoans and handling cell destiny selections in vast number of stem cell lineages [5, 6]. In this study, we have taken into consideration of prominent plant components with abundant therapeutic properties. Although many researches were accomplished on those substances up to now, their molecular dynamics has not been studied. One of them is nigellin-1.1, and it is classified as an antimicrobial protein. Nigellin-1.1 is a novel type of hairpin-like defense peptides with three disulfide bridges from blackseed (Nigella sativa L., NS), a member of the Ranunculaceae (Buttercup family). NS has a dark brown or black in color with a distinguished angular (funnel-shape). It has a strong aroma and little bitter nutty-peppery taste [7, 8]. NS has been used since traditionally and now, when there was a need to develop a scientific concept on the use of herbal remedies in the treatment of human diseases and relief from their suffering. Regulation of reactive species interfering with DNA structure, modulating various potential targets and their signaling pathways as well as immunomodulatory effects, anticancer, antitussive, antidiabetic, antioxidant, gastroprotective, neuro-protective, hepatoprotective, immunomodulator, analgesic, spasmolytic, anti-inflammatory, antimicrobial, and bronchodilator activity were confirmed by a lot of pharmacological in-vitro and in-vivo studies on NS [9, 10]. Research in 2017 reported Nigella sativa seed management on prevention of febrile neutropenia all through chemotherapy among youngsters with brain tumors [11]. Another studied ingredient is curcumin. Curcumin is a hydrophobic polyphenol extracted from dried rhizome of turmeric (Curcuma longa), which is a member of the ginger family. A chemical name of curcumin is “Diferuloylmethane” with the C21H20O6 chemical formula [12, 13]. Curcumin’s effects on glioma cells in vitro and in vivo were properly studied. Curcumin has many molecular objectives and consequently numerous and complicated mechanisms of action. The antitumoral consequences of curcumin are the notion to behave through many exclusive signaling pathways inclusive of cell proliferation, angiogenesis, immunomodulation, metastasis, autophagy, invasion, and apoptosis [14]. In addition to its effectiveness as an antineoplastic, curcumin has been observed to be defensive toward reactive oxygen species (ROS) [15, 16]. Data additionally exists to signify that curcumin acts synergistically with chemotherapeutics already permitted for the remedy of brain tumors. For example, while doxorubicin was added in combination with 25 μmol/L curcumin, the viability dropped to 36% and 46% for both U138 and C6 GBM cell lines (Glioblastoma (GBM) is the most common malignant primary CNS tumor), respectively [17]. In 2016, Cui et al. found that the combined drugs (paclitaxel and curcumin) yielded synergistic effects on inhibition of brain tumor growth through the mechanisms of cell cycle arrest and apoptosis induction. Moreover, outcomes exhibited significantly enhancement in efficacy of drugs and chemotherapy. [18]. A computer simulation is the presentation of the dynamic reactions of one system by the behavior of another system patterned after it. A simulation employs a model or mathematical description of a natural system in the form of a computer program. This model is composed of equations that replicate the functional connections seen in the existing system. When the program is run, the ensuing mathematical dynamics constitutes an analog of the current system's behavior, and the outcomes are represented as data. The most significant computer simulation capable of predicting the atomic behavior of a range of structures is the Molecular Dynamics (MD) approach [19, 20]. This computational technique is now frequently employed in modeling living structures [21]. Amongst the neurodegenerative illnesses, one of the most common ones is Alzheimer's disease (AD) determined through progressive accumulation of amyloid-beta (Aβ) peptides in brain. Based on research in 2018, by the use of MD simulations, the conformational transitions and molecular interactions in the presence of curcumin supplied further insight into the structural information of Aβ for designing of effective inhibitors towards amyloid aggregation [22]. In another study, a series of molecular dynamics simulations had been carried out in order to provide an explanation for the underlying molecular mechanism of the destabilization of Aβ protofibrils through curcumin, carmustine, acyclovir, and tetracycline. It was found from outcomes that each one of these drugs, including curcumin, attach to the interior of the hydrophobic grooves of Aβ protofibrils and caused destabilization of the β-strand of Aβ protofibrils [23]. In new study, the MD simulation results represented that Aβ interaction with Aspirin was stable in water system when placed within Cu nanochannels. After a time, interval of t = 10 ns, the highest velocity and temperature characteristics /density, exceed 17.73 Å/fs, 1451.33 K, and 0.1426 atoms/Å3, respectively [24, 25]. Iron deposition in the central nervous system (CNS) is one of the reasons of neurodegenerative illnesses. Human transferrin (hTf) operates as an iron carrier present in the blood plasma, inhibiting it from contributing to redox reactions. A 250 ns molecular dynamics simulation has been carried out which deciphered the dynamics and stability of the hTf-TQ complex. Thymoquinone (TQ) is the main active ingredient of blackseed. Structure analysis offered that the binding of TQ doesn't reason any significant transformations in the hTf structure within the course of simulation and a stable complex was formed [26]. Also, black seed oil-induced inhibitory results were associated with a considerable diminution in mRNA expression of epigenetic complex including UHRF1, DNMT1 and HDAC1. Molecular docking and MD simulation indicated that TQ in blackseed oil had excellence binding affinity to UHRF1 and HDAC1, and it could inhibit them [27]. Theoretical calculations were done in this study to anticipate the effects of curcumin and nigellin-1.1 on brain antitumor interactions at different temperatures (300–350 K). The temperature-dependent dynamical way of atomic structures can be predicted using the equilibrating molecular dynamics approach, which regulates the temperature of the atomic structure.
Computational Methods
The MD approach was utilized to assessing the atomic interactions of curcumin and nigellin-1.1 on brain antitumor molecule at temperature intervals of 300 K–350 K. In this work, 21181 atoms were used, and the grid size was set to 50 × 42 × 50. All units are based on Angstroms. Conjugate Gradient method was utilized for optimization [28]. MD is used to identify the physical movement of molecules and atoms. The atomic interaction of curcumin and nigellin-1.1 on brain antitumor may be caused chemically by a lack of hydrogen in these structures. The free bond is detected, and adsorption sites are produced between these two structures as a result of this atomic mechanism. Figure 1 shows the structures of curcumin (3OV2) and nigellin-1.1 (2NB2) as well as a brain antitumor (1Q7F) [29, 30].

Fig. 1 Schematic of (a) curcumin (3OV2), (b) nigellin-1.1 (2NB2), and (c) brain antitumor (1Q7F)
Particle interactions are enabled for a set amount of time (time step), allowing for a better understanding of the dynamical development of the simulated particles. The trajectories of particles are calculated in the most common manner by computationally solving Newton's equations of motion for a system of particles, where the forces between the particles are computed by interatomic force-field [31]. The current MD simulation was carried out using the LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) program [32–36]. This MD tool was created by Sandia National Laboratories (SNL). LAMMPS provides us with a number of options.
Step 1: Atomic details were simulated individually for curcumin, nigellin-1.1, and brain antitumor components. In the following MD computations, the simulation box has a length of 100 in the x, y, and z dimensions, with a periodic boundary condition in each of these directions [37]. The temperature of the atomic structures was equilibrated at 300 K, 325 K, and 350 K with a 1 ns time step in the following MD simulations. The Nose-Hoover thermostat with 0.01 damping rate for 1 ns was employed in the temperature equilibration procedure [38–41]. This rate of temperature damping is consistent with the studies of Ashkezari et al[42]. The total energy of simulated structures was given in this stage to ensure that the atomic model and interatomic force field were correct. The computational running was then maintained for 2 ns after the system attained equilibrium temperature and overall energy rate.
Step 2: Curcumin and brain antitumor molecule, as well as nigellin-1.1 and brain antitumor molecule, were simulated in a similar simulation box in the second step. At 300 K, 325 K, and 350 K, the simulated structures were equilibrated for 1 ns. Finally, a 2 ns atomic interaction between curcumin and nigellin-1.1 was performed on brain antitumor structures. Physical metrics such as total energy, structure distance, diffusion coefficient, and volume of brain antitumor after contacting with curcumin and nigellin-1.1 structures were reported to analyze the atomic interaction between these two structures.
The interatomic force between different atoms in curcumin, nigellin-1.1, and brain antitumor is handled for in the current MD simulations using the Universal Force Field (UFF) and DREIDING force-field [43, 44]. DREIDING and UFF force-fields were created by Goddard et al. The DREIDING force-field was designed to predict the structures and dynamics of organic, biological, and main-group inorganic molecules. UFF also has settings for each atom. The force-field parameters are computed using generic criteria based merely on the element, its hybridization, and its connectedness. The primary principle of DREIDING and UFF force-fields is to employ generic force constants and geometrical parameters based on simple hybridization considerations. For non-bond interaction between distinct atoms, the LJ potential is used in several force fields [45]:
![]() (1)
(1)
The depth of the potential well is denoted by ε, the finite distance at which the interatomic potential is zero is denoted by σ, and the distance between the atoms is denoted by r. The cutoff radius is represented by rc in this equation, which accounts for 12 Å in Eq.(1). Table 1 shows the ε and σ constants for several simulated atoms.
Table 1 MD simulation duration and energy parameters for the LJ potential [43, 44]
Atom | σ (Å) | ε (kcal/mol) |
C | 4.180 | 0.305 |
N | 3.995 | 0.415 |
S | 2.240 | 0.305 |
H | 3.200 | 0.010 |
O | 3.710 | 0.415 |
Harmonious relation is also employed in DREIDING and UFF force-fields for bond and angle interactions [36]:
![]() (2)
(2)
![]() (3)
(3)
The harmonic constant is kr, and the equilibrium distance of atomic bonds is r0. In Eq. (3), kθ is the angular harmonic constant, and θ0 is the angle bond equilibrium angle. Tables 2 and 3 show the constant rate for simulated atomic structures in this study.
Table 2 The harmonic constants and equilibrium distances for the atomic structures in the simulation box [38, 39]
Bond | kr (kcal/mol) | r0 (Å) |
C–C | 700 | 1.530 |
C–N | 700 | 1.462 |
C–O | 700 | 1.420 |
C–S | 700 | 1.800 |
H–O | 700 | 0.980 |
C–H | 700 | 1.090 |
H–N | 700 | 1.022 |
S–S | 700 | 2.070 |
Table 3 The angular harmonic constant and equilibrium degree for atomic structures in the simulation box
Angle | kθ (kcal/mol) | θ0 |
C–C–C | 100 | 109.471 |
C–C–N | 100 | 109.471 |
C–C–S | 100 | 109.471 |
C–C–O | 100 | 109.471 |
C–N–C | 100 | 106.700 |
C–S–C | 100 | 92.100 |
H–O–H | 100 | 104.510 |
H–C–H | 100 | 109.471 |
N–C–O | 100 | 109.471 |
O–C–O | 100 | 109.471 |
At the atomic level, Newton's second law has been employed as the gradient of the potential function for calculations of particle motion over time, as seen in the equations [46]:
![]() (4)
(4)
![]() (5)
(5)
where Fi is the total force, mi is the atomic mass, ri is the position, and Vi is the velocity of atom i in this equation. The forces between particles are computed in MD simulations using interatomic potential.
Results and Discussion
Equilibration of atomic structures
Curcumin, nigellin-1.1, and brain antitumor atomic structures were investigated in this portion, and the final particle positions were stored for the following step of MD simulation (Fig. 2). Open Visualization Tool (OVITO) software [47] was used to display the atomic structure figures given in this work. For atomistic and particle-based simulation data, OVITO is a scientific visualization and analysis program. We determine the potential energy for isolated structures of curcumin, nigellin-1.1, and brain antitumor after atomic modeling. This physical parameter converged to 1200 eV and 1054 eV after 1 ns at room temperature for curcumin and brain antitumor structures, and to 931 eV and 815 eV after 1 ns at room temperature for nigellin-1.1 and brain antitumor structures. The atomic stability of these two structures is shown in this way. Physically, these MD simulations revealed that using UFF and DREIDING force-fields, the starting location of atoms in curcumin, nigellin-1.1, and brain antitumor particles described in prior studies is adopted. The total energy of these atomic structures as a function of sample temperatures is used to indicate the stability of atomic structures.

Fig. 2 Atomic representations of (a) the curcumin, (b) the nigellin-1.1 and (c) brain antitumor simulated in NVT ensemble using the LAMMPS software
This method is used to increase the kinetic energy of atoms in atomic structure. The mean distance of atoms rose when the kinetic energy was raised. Potential energy reciprocal interactions with a mean distance of atoms exist physically, and as the mean distance of atoms increases, so does the potential and total energy of atomic structures. The atomic stability reduces when this physical property lowers. Furthermore, as previously stated [21, 42], the results of this computational phase reveal that UFF and DREIDING force fields have high ability in bio-structures.
These atomic structures were simulated in a single box to explore the atomic interaction between curcumin, nigellin-1.1, and brain antitumor. Each molecule's equilibrated atomic structure was retrieved in the preceding phase. The MD simulation was run for 1 ns after the atom simulation to achieve thermal equilibration of the structures. Figures 3 and 4 show the starting placements of the two structures. The initial distance between curcumin and brain antitumor, and nigellin-1.1 and brain antitumor, in this atomic combination, is smaller than the cutoff radius for modeling of molecular interaction. Figure 5 shows the total energy variation of curcumin and brain antitumor, as well as nigellin-1.1 and brain antitumor. These statistics show that the overall energy of the final structure is greater than the energy of each individual structure. The total energy rate has a direct relationship with the stability of atomic structures. As a result, the rise in total energy in this stage of MD simulations indicates that curcumin and brain antitumor, as well as nigellin-1.1 and brain antitumor combination, are stable.
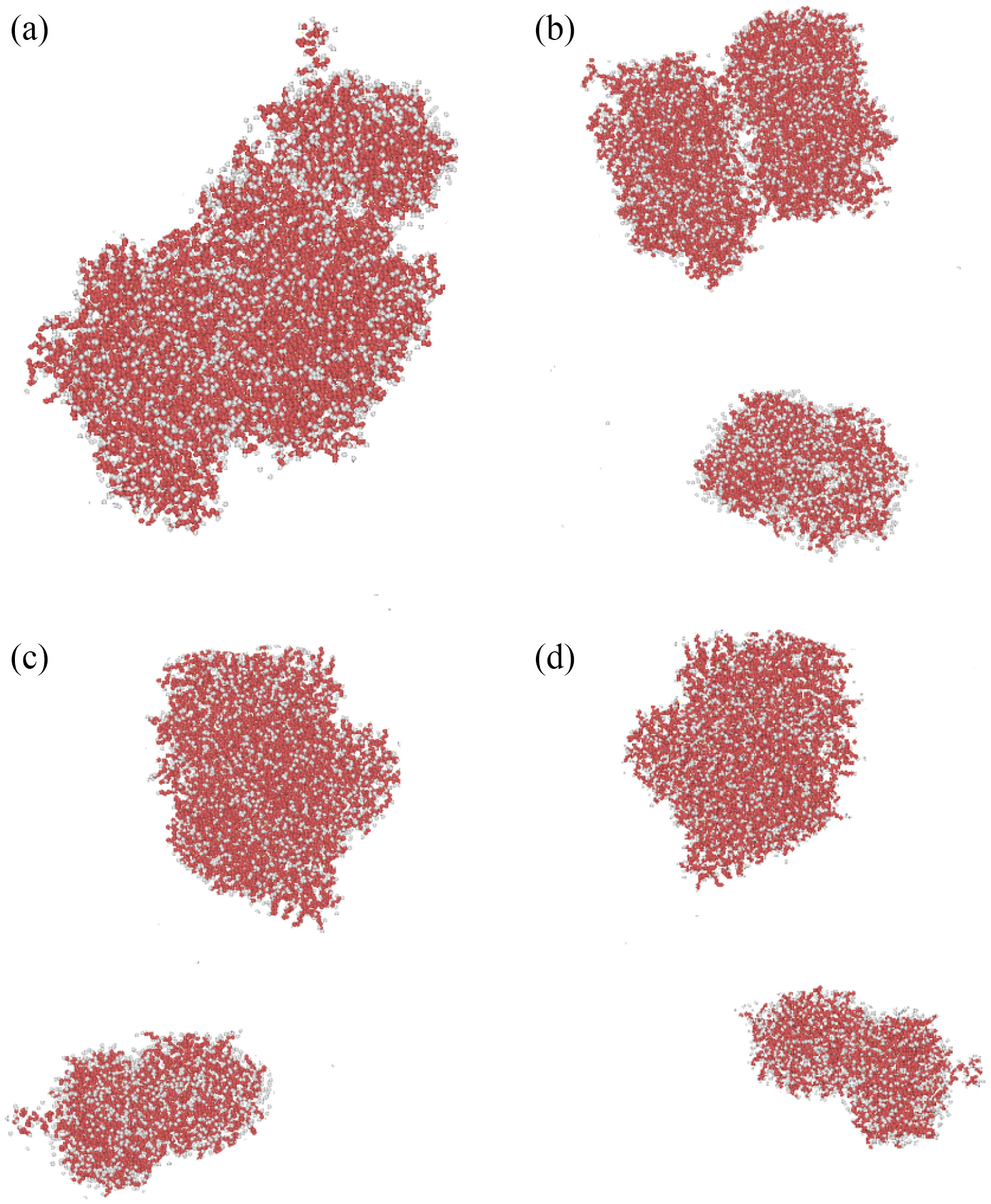
Fig. 3 Atomic representations of simulated curcumin and brain antitumor molecular mixes using the LAMMPS software at (a) the top, (b) the front, (c) the left, and (d) the perspective viewpoints

Fig. 4 Atomic representations of simulated nigellin-1.1 and brain antitumor molecular mixes using the LAMMPS software at (a) the top, (b) the front, (c) the left, and (d) the perspective viewpoints

Fig. 5 The total energy variation of (a) nigellin-1.1 and brain antitumor and (b) curcumin and brain antitumor molecule as a function of simulation time and sample temperatures
Atomic structure dynamical evolution
Canonical ensemble proceeded for another 2 ns after initial atomic MD simulations and temperature equilibration of curcumin and brain antitumor, as well as nigellin-1.1 and brain antitumor. A statistical ensemble known as the canonical ensemble represents the potential states of a mechanical system in thermal equilibrium with a heat bath at a constant temperature in statistical mechanics.
The energy exchange between the system and the heat bath results in a difference in the system's total energy states. The center of mass (COM) distance between two atomic structures was reported in the first phase of this section. The unique location where the weighted relative position of the dispersed mass accumulates to zero is the center of mass of a distribution of mass in space [48]. We may deduce from Fig. 6 that the interatomic force is attractive. At 300 K, the distance between curcumin and brain antitumor molecule ranges between 1.16 Å and 1.12 Å, whereas the distance between nigellin-1.1 and brain antitumor molecule varies between 2.01 Å and 1.93 Å. As the temperature rises, this value falls (Fig. 6, Table 4 and Table 5). The amplitude of the atomic fluctuations is increased, resulting in this kind of atomic structure. Physically, increasing atomic fluctuations reduces the attraction force between atoms, resulting in more free space between them. As a result, raising the amplitude of atomic fluctuation leads more atoms to penetrate at the same time.
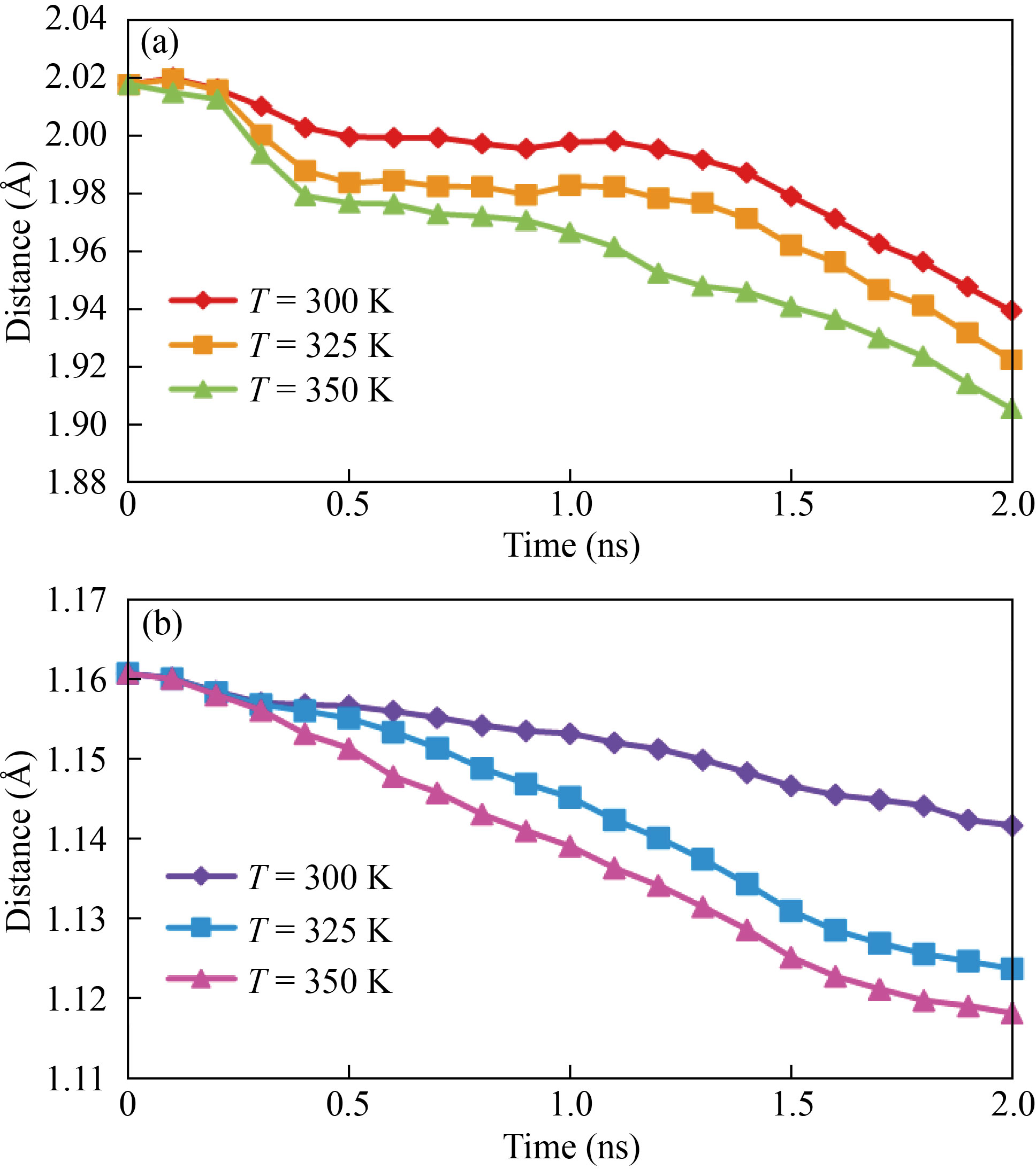
Fig. 6 The fluctuation in COM distance between atomic structures of (a) nigellin-1.1 and brain antitumor and (b) curcumin and brain antitumor as a function of simulation time and sample temperature
Table 4 The COM distance between nigellin-1.1 and brain antitumor atomic structures fluctuates with sample temperature
Temperature (K) | COM distance (Å) |
300 | 1.935 |
325 | 1.921 |
350 | 1.901 |
Table 5 The COM distance between curcumin and brain antitumor atomic structures fluctuates with sample temperature
Temperature (K) | COM distance (Å) |
300 | 1.141 |
325 | 1.123 |
350 | 1.110 |
In MD simulations, the diffusion coefficient is the optimum parameter for understanding atomic interactions [49]. The diffusion coefficient was estimated in the LAMMPS program, and the mean-squared displacement (MSD) of the system was obtained using the "compute msd" command in this stage of the MD simulations. The diffusion coefficient is proportional to the slope of the MSD vs simulation time.
The data reveals that raising the temperature raises the diffusion coefficient from 0.89 µm2/s to 0.93 µm2/s and 0.36 µm2/s to 0.41 µm2/s for curcumin and brain antitumor and nigellin-1.1 and brain antitumor, respectively, by temperature ranging from 300 K to 350 K (see Tables 6 and 7). The decrease in the potential and total energy of these structures causes an increase in the amplitude of atomic fluctuation, resulting in an increase in the physical parameter.
Table 6 The diffusion coefficient of nigellin-1.1 and brain antitumor atomic structures varies as a function of sample temperature
Temperature (K) | Diffusion coefficient (µm2/s) |
300 | 0.36 |
325 | 0.38 |
350 | 0.41 |
Table 7 The diffusion coefficient of curcumin and brain antitumor atomic structures varies as a function of sample temperature
Temperature (K) | Diffusion coefficient (µm2/s) |
300 | 0.89 |
325 | 0.91 |
350 | 0.93 |
In MD simulations, as the diffusion coefficient rises, the diffusion of curcumin and nigellin-1.1 into brain antitumor increases, lowering the stability of the molecule and destroying the atomic structure. Our computed physical parameters in this phase of the MD research reveal that changing the temperature of simulated structures affects their atomic behavior. Thus, in real-life situations, temperature limitation may lessen the damaging consequences of a brain antitumor and shorten the treatment duration.
Because the atomic volume of each atomic structure is related to its stability, brain antitumor molecule volume fluctuation as a function of simulation duration from 0 to 2 ns demonstrates this structure stability. The current simulations reveal that the volume of the brain antitumor molecule increases when time steps are increased. The atomic distance rises at a faster pace as the molecule's volume is increased [49–53]. The overall energy of a molecule falls as the distance between atoms increases, and the stability of the atomic structure diminishes. The amount of brain antitumor interaction with curcumin grows from 1.33×106 Å3 to 2.83×106 Å3 as the MD simulation time progresses from 0 to 2 ns, and the volume of brain antitumor interaction with nigellin-1.1 climbs from 1.33×106 Å3 to 2.24×106 Å3. The ultimate volume of brain antitumor interaction with curcumin and brain antitumor interaction with nigellin-1.1 changes from 2.83×106 to 2.88×106 Å3 and 2.24×106 Å3 to 2.69×106 Å3 correspondingly when the temperature rises from 300 K to 350 K. Finally, it can be deduced that the drop in total energy in this stage of MD simulation is due to a decrease in the brain antitumor's stability. The volume of brain antitumor contact with curcumin and brain antitumor interaction with nigellin-1.1 structures are shown schematically in Fig. 7, Fig. 8 and Table 8. The "construct surface mesh" feature of the OVITO program was used to create these figures and calculate their volumes (Fig. 9).
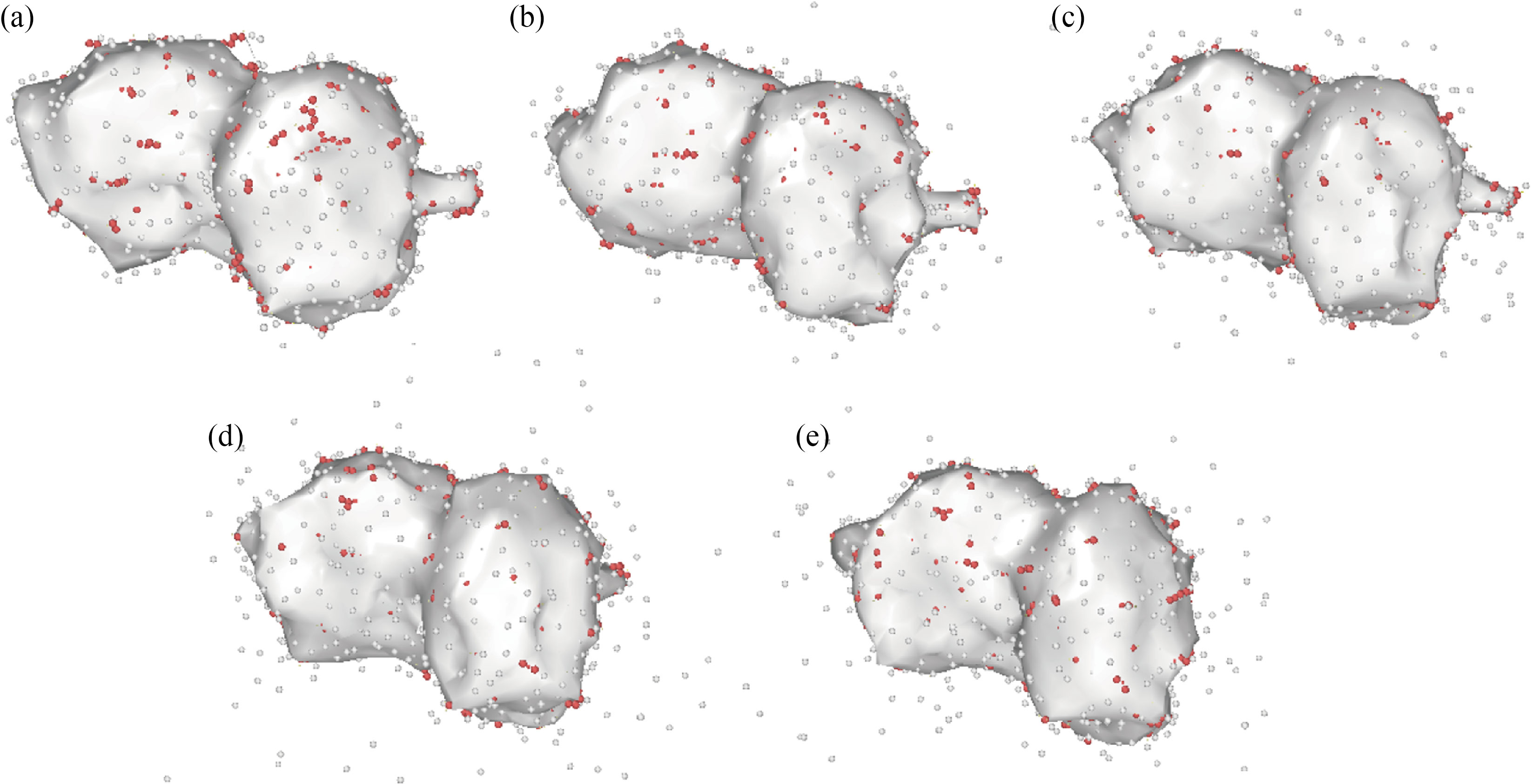
Fig. 7 The volume of the brain antitumor interaction with curcumin atomic structure changes as the MD simulation time increases
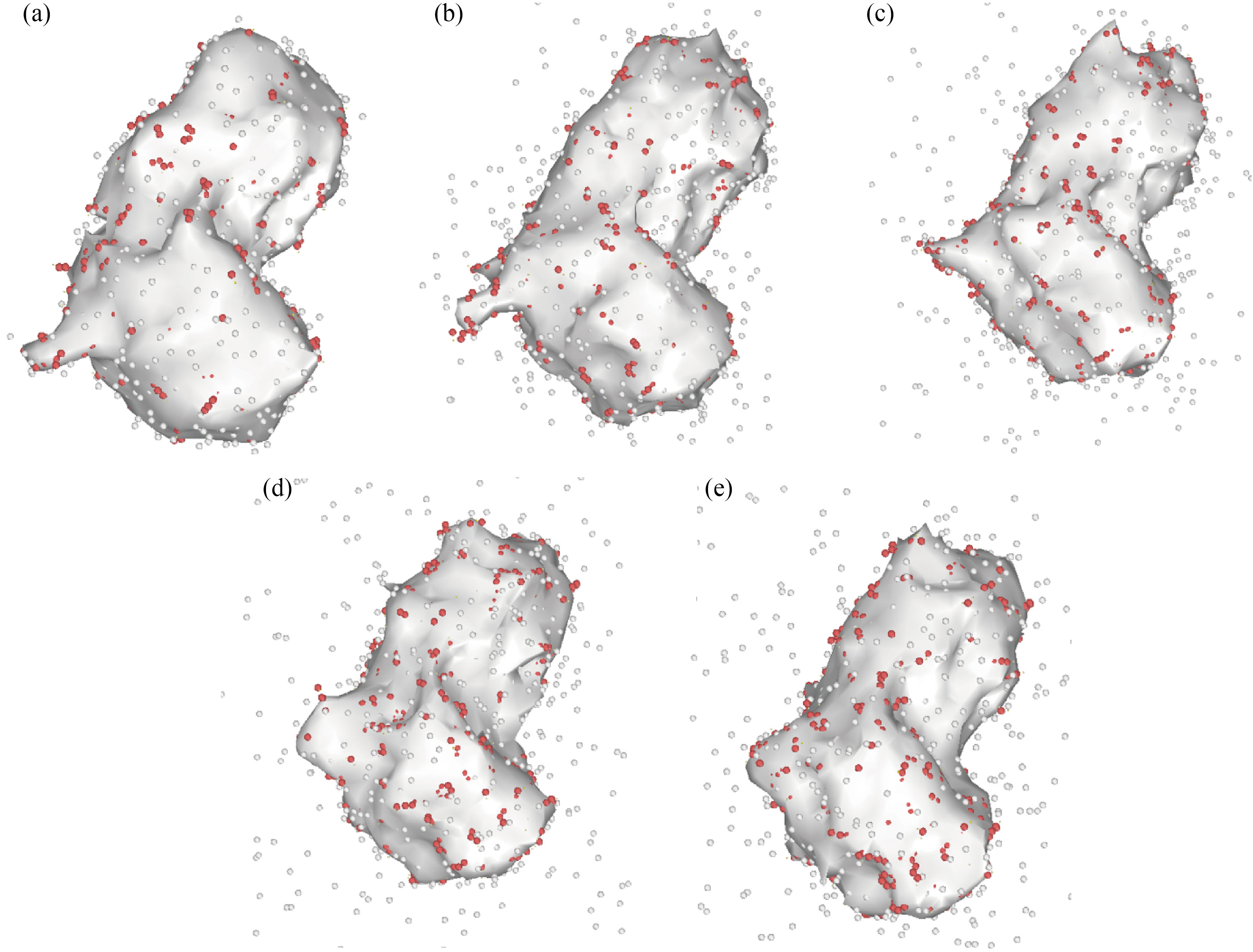
Fig. 8 The volume of the brain antitumor interaction with nigellin-1.1 atomic structure changes as the MD simulation time increases
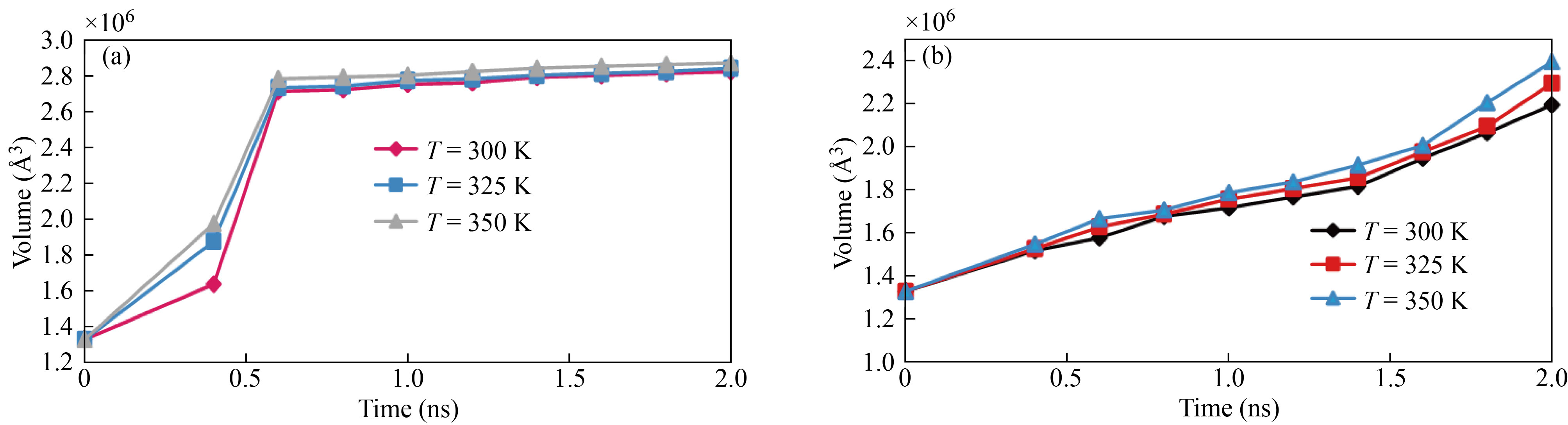
Fig. 9 Time development of (a) nigellin-1.1 and brain antitumor volume and (b) curcumin and brain antitumor volume ss a function of temperature
Table 8 Volume variation of brain antitumor interaction with curcumin and nigellin-1.1 as a function of sample temperature
Temperature (K) | Brain antitumor interaction with curcumin volume (Å3) | Brain antitumor interaction with nigellin-1.1 volume (Å3) |
300 | 2.245 | 2.831 |
325 | 2.470 | 2.845 |
350 | 2.695 | 2.885 |
Conclusion
The influence of curcumin and nigellin-1.1 on the atomic organization of brain antitumor is investigated in this research. C, H, N, O, and S atoms are used to represent each curcumin, nigellin-1.1, and brain antitumor in this approach. UFF and DREIDING force-fields have also been created to imitate the interatomic force. Finally, here are the computational findings from MD simulations:
(1) For curcumin and brain antitumor structures converged to 1054 eV and for nigellin-1.1 and brain antitumor structures converged to 815 eV after 1 ns at 300 K, UFF and DREIDING force-fields are the ideal interatomic potential for energy, and this atomic approach illustrates the stability of these structures.
(2) In MD simulations, the center of mass for curcumin and brain antitumor, as well as nigellin-1.1 and brain antitumor, decreases from 1.16 Å to 1.12 Å and 2.01 Å to 1.93 Å, respectively, as time passes.
(3) For curcumin and brain antitumor and nigellin-1.1 and brain antitumor, raising the atomic temperature from 300 K to 350 K increases the diffusion coefficient from 0.89 μm2/s to 0.93 μm2/s and 0.36 μm2/s to 0.41 μm2/s, respectively.
(4) By incorporating more curcumin and nigellin-1.1 into the atomic structure of brain antitumor, the stability of the antitumor is increased.
(5) Diffusion of curcumin and nigellin-1.1 into this atomic structure increases the volume of the brain antitumor.
From a practical standpoint, we conclude that managing the temperature of curcumin and nigellin-1.1 and the brain antitumor is a key parameter in real instances to decrease the harmful consequences of brain antitumor.
References
[1] P. Zhang, F. Wang, J.Y. Hu, et al. Exploring the relationship between drug side-effects and therapeutic indications. AMIA Annual Symposium Proceedings Archive, 2013, 2013: 1568–1577. https://pubmed.ncbi.nlm.nih.gov/24551427
[2] S.I. Ngulde, U.K. Sandabe, R. Abounader, et al. Activities of some medicinal plants on the proliferation and invasion of brain tumor cell lines. Advances in Pharmacological and Pharmaceutical Sciences, 2020, 2020: 3626879. https://doi.org/10.1155/2020/3626879
[3] F. Akter, B. Simon, N.L. de Boer, et al. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer, 2021, 1875: 188458. http://dx.doi.org/10.1016/j.bbcan.2020.188458
[4] F. Hirth. Drosophila melanogaster in the study of human neurodegeneration. CNS & Neurological Disorders - Drug Targets, 2010, 9: 504–523. https://doi.org/10.2174/187152710791556104
[5] I. Loedige, L. Jakob, T. Treiber, et al. The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of Drosophila brain-tumor-mediated gene regulation. Cell Reports, 2015, 13: 1206–1220. http://dx.doi.org/10.1016/j.celrep.2015.09.068
[6] P. Climent-Cantó, A. Carbonell, S. Tamirisa, et al. The tumour suppressor brain tumour (Brat) regulates linker histone dBigH1 expression in the Drosophila female germline and the early embryo. Open Biology, 2021, 11: 200408. https://doi.org/10.1098/rsob.200408
[7] K. Kotecka-Majchrzak, A. Sumara, E. Fornal, et al. Identification of species-specific peptide markers in cold-pressed oils. Scientific Reports, 2020, 10: 19971. http://dx.doi.org/10.1038/s41598-020-76944-z
[8] P. Sharma, T. Longvah. Nigella (Nigella sativa) Seed. Oilseeds: Health Attributes and Food Applications. Singapore: Springer, 2021: 331–350. https://doi.org/10.1007/978-981-15-4194-0_13
[9] M. Faruque, Ahmad, . An updated knowledge of Black seed (Nigella sativa Linn.): Review of phytochemical constituents and pharmacological properties. Journal of Herbal Medicine, 2021, 25: 100404. http://dx.doi.org/10.1016/j.hermed.2020.100404
[10] C.H. Phua, Z.L. Teoh, B.H. Goh, et al. Triangulating the pharmacological properties of thymoquinone in regulating reactive oxygen species, inflammation, and cancer: Therapeutic applications and mechanistic pathways. Life Sciences, 2021, 287: 120120. https://doi.org/10.1016/j.lfs.2021.120120
[11] H.F.M. Mousa, N.K. Abd-El-Fatah, O.A.H. Darwish, et al. Effect of Nigella sativa seed administration on prevention of febrile neutropenia during chemotherapy among children with brain tumors. Child's Nervous System, 2017, 33: 793–800. http://dx.doi.org/10.1007/s00381-017-3372-7
[12] M. Modaresi, M. HarfBol, F. Ahmadi. A review on pharmacological effects and therapeutic properties of curcumin. Journal of Medicinal Plants, 2017, 16(62): 1–17. http://dorl.net/dor/20.1001.1.2717204.2017.16.62.10.3
[13] V. Zoi, V. Galani, G.D. Lianos, et al. The role of curcumin in cancer treatment. Biomedicines, 2021, 9: 1086. https://doi.org/10.3390/biomedicines9091086
[14] A.K. Khaw, M.P. Hande, G. Kalthur, et al. Curcumin inhibits telomerase and induces telomere shortening and apoptosis in brain tumour cells. Journal of Cellular Biochemistry, 2013, 114: 1257–1270. https://doi.org/10.1002/jcb.24466
[15] A. Karthikeyan, N. Senthil, T. Min. Nanocurcumin: A promising candidate for therapeutic applications. Frontiers in Pharmacology, 2020, 11: 487. https://doi.org/10.3389/fphar.2020.00487
[16] N.V. Klinger, S. Mittal. Therapeutic potential of curcumin for the treatment of brain tumors. Oxidative Mmedicine and Cellular Longevity, 2016, 2016: 9324085. https://doi.org/10.3389/fphar.2020.00487
[17] A. Zanotto-Filho, E. Braganhol, M.I. Edelweiss, et al. The curry spice curcumin selectively inhibits cancer cells growth in vitro and in preclinical model of glioblastoma. Journal of Nutritional Biochemistry, 2012, 23: 591–601. https://doi.org/10.1016/j.jnutbio.2011.02.015
[18] Y.N. Cui, M. Zhang, F. Zeng, et al. Dual-targeting magnetic PLGA nanoparticles for codelivery of paclitaxel and curcumin for brain tumor therapy. ACS Applied Materials & Interfaces, 2016, 8: 32159–32169. https://doi.org/10.1021/acsami.6b10175
[19] R. Maleki, H.H. Afrouzi, M. Hosseini, et al. Molecular dynamics simulation of Doxorubicin loading with N-isopropyl acrylamide carbon nanotube in a drug delivery system. Computer Methods and Programs in Biomedicine, 2020, 184: 105303. http://dx.doi.org/10.1016/j.cmpb.2019.105303
[20] M. Tohidi, D. Toghraie. The effect of geometrical parameters, roughness and the number of nanoparticles on the self-diffusion coefficient in Couette flow in a nanochannel by using of molecular dynamics simulation. Physica B: Condensed Matter, 2017, 518: 20–32. http://dx.doi.org/10.1016/j.physb.2017.05.014
[21] N.A. Jolfaei, N.A. Jolfaei, M. Hekmatifar, et al. Investigation of thermal properties of DNA structure with precise atomic arrangement via equilibrium and non-equilibrium molecular dynamics approaches. Computer Methods and Programs in Biomedicine, 2020, 185: 105169. http://dx.doi.org/10.1016/j.cmpb.2019.105169
[22] M. Awasthi, S. Singh, V.P. Pandey, et al. Modulation in the conformational and stability attributes of the Alzheimer’s disease associated amyloid-beta mutants and their favorable stabilization by curcumin: Molecular dynamics simulation analysis. Journal of Biomolecular Structure and Dynamics, 2018, 36: 407–422. http://dx.doi.org/10.1080/07391102.2017.1279078
[23] I. Jahan, S.M. Nayeem. Destabilization of Alzheimer’s Aβ42 protofibrils with acyclovir, carmustine, curcumin, and tetracycline: Insights from molecular dynamics simulations. New Journal of Chemistry, 2021, 45: 21031–21048. https://doi.org/10.1039/d1nj04453b
[24] Q.H. Le, S. Heydaripour, B. Farhadi, et al. A molecular dynamics investigation of the atomic structural behavior of the interaction between amyloid beta protein and Aspirin within an aqueous nanodomain. Engineering Analysis with Boundary Elements, 2023, 146: 851–858. http://dx.doi.org/10.1016/j.enganabound.2022.11.018
[25] Z. Karimi, S. Rahmani, B. Farhadi, et al. Synthesis of 4-arylidene-1-(4-phenylselenazol-2-yl)-3-methyl-1H-pyrazol-5(4H)-ones via a four-component condensation. Polycyclic Aromatic Compounds, 2022, 69: 1–7. https://doi.org/10.1080/10406638.2022.2149570
[26] B. Xue, D. DasGupta, M. Alam, et al. Investigating binding mechanism of thymoquinone to human transferrin, targeting Alzheimer’s disease therapy. Journal of Cellular Biochemistry, 2022, 123: 1381–1393. https://doi.org/10.1002/jcb.30299
[27] S. Alsanosi, R.A. Sheikh, S. Sonbul, et al. The potential role of Nigella sativa seed oil as epigenetic therapy of cancer. Molecules, 2022, 27: 2779. https://doi.org/10.3390/molecules27092779
[28] S.J. Watowich, E.S. Meyer, R. Hagstrom, et al. A stable, rapidly converging conjugate gradient method for energy minimization. Journal of Computational Chemistry, 1988, 9: 650–661. https://doi.org/10.1002/jcc.540090611
[29] T.A. Edwards, B.D. Wilkinson, R.P. Wharton, et al. Model of the brain tumor-Pumilio translation repressor complex. Genes & Development, 2003, 17: 2508–2513. https://doi.org/10.1101/gad.1119403
[30] R. Satou, A. Miyanaga, H. Ozawa, et al. Structural basis for cyclization specificity of two Azotobacter type III polyketide synthases: A single amino acid substitution reverses their cyclization specificity. Journal of Biological Chemistry, 2013, 288: 34146–34157. https://doi.org/10.1074/jbc.m113.487272
[31] L.P. Wang, T.J. Martinez, V.S. Pande. Building force fields: An automatic, systematic, and reproducible approach. The Journal of Physical Chemistry Letters, 2014, 5: 1885–1891. https://doi.org/10.1021/jz500737m
[32] S. Plimpton. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics, 1995, 117: 1–19. http://dx.doi.org/10.1006/jcph.1995.1039
[33] S.J. Plimpton, A.P. Thompson. Computational aspects of many-body potentials. MRS Bulletin, 2012, 37: 513–521. http://dx.doi.org/10.1557/mrs.2012.96
[34] W.M. Brown, P. Wang, S.J. Plimpton, et al. Implementing molecular dynamics on hybrid high performance computers - short range forces. Computer Physics Communications, 2011, 182: 898–911. http://dx.doi.org/10.1016/j.cpc.2010.12.021
[35] W.M. Brown, A. Kohlmeyer, S.J. Plimpton, et al. Implementing molecular dynamics on hybrid high performance computers - Particle-particle particle-mesh. Computer Physics Communications, 2012, 183: 449–459. http://dx.doi.org/10.1016/j.cpc.2011.10.012
[36] B. Farhadi, F. Zabihi, S.Y. Yang, et al. Carbon doped lead-free perovskite with superior mechanical and thermal stability. Molecular Physics, 2022, 120: e2013555. http://dx.doi.org/10.1080/00268976.2021.2013555
[37] B. Farhadi, M. Ciprian, F. Zabihi, et al. Influence of contact electrode and light power on the efficiency of tandem perovskite solar cell: Numerical simulation. Solar Energy, 2021, 226: 161–172. http://dx.doi.org/10.1016/j.solener.2021.08.043
[38] S. Nosé. A unified formulation of the constant temperature molecular dynamics methods. The Journal of Chemical Physics, 1984, 81: 511–519. http://dx.doi.org/10.1063/1.447334
[39] W.G. Hoover. Canonical dynamics: Equilibrium phase-space distributions. Physical Review A, 1985, 31: 1695–1697. https://doi.org/10.1103/physreva.31.1695
[40] W. Humphrey, A. Dalke, K. Schulten. VMD: Visual molecular dynamics. Journal of Molecular Graphics, 1996, 14: 33–38. https://doi.org/10.1016/0263-7855(96)00018-5
[41] M. Ibrahim, T. Saeed, M. Hekmatifar, et al. Investigation of dynamical behavior of 3LPT protein - water molecules interactions in atomic structures using molecular dynamics simulation. Journal of Molecular Liquids, 2021, 329: 115615. http://dx.doi.org/10.1016/j.molliq.2021.115615
[42] A.Z. Ashkezari, N.A. Jolfaei, N.A. Jolfaei, et al. Calculation of the thermal conductivity of human serum albumin (HSA) with equilibrium/non-equilibrium molecular dynamics approaches. Computer Methods and Programs in Biomedicine, 2020, 188: 105256. http://dx.doi.org/10.1016/j.cmpb.2019.105256
[43] A.K. Rappe, C.J. Casewit, K.S. Colwell, et al. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. Journal of the American Chemical Society, 1992, 114: 10024–10035. https://doi.org/10.1021/ja00051a040
[44] S.L. Mayo, B.D. Olafson, W.A. Goddard. DREIDING: A generic force field for molecular simulations. Journal of Physical Chemistry, 1990, 94: 8897–8909. https://doi.org/10.1021/j100389a010
[45] J.E. Jones. On the determination of molecular fields.—II. From the equation of state of a gas. Proceedings of the Royal Society A, 1924, 106: 463–477. http://dx.doi.org/10.1098/RSPA.1924.0082
[46] A. Karimipour, A. Karimipour, N.A. Jolfaei, et al. Prediction of the interaction between HIV viruses and Human Serum Albumin (HSA) molecules using an equilibrium dynamics simulation program for application in bio medical science. Journal of Molecular Liquids, 2020, 318: 113989. http://dx.doi.org/10.1016/j.molliq.2020.113989
[47] Stukowski. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Modelling and Simulation in Materials Science and Engineering, 2010, 18: 015012. https://doi.org/10.1088/0965-0393/18/1/015012
[48] R.P. Feynman, R.B. Leighton, M. Sands. The new millennium edition: Mainly mechanics, radiation, and heat. The Feynman lectures on physics. New York: Basic Books, 2011.
[49] Y. Orooji, R. Mohassel, O. Amiri, et al. Gd2ZnMnO6/ZnO nanocomposites: Green sol-gel auto-combustion synthesis, characterization and photocatalytic degradation of different dye pollutants in water. Journal of Alloys and Compounds, 2020, 835: 155240. http://dx.doi.org/10.1016/j.jallcom.2020.155240
[50] Y. Orooji, M. Ghanbari, O. Amiri, et al. Facile fabrication of silver iodide/graphitic carbon nitride nanocomposites by notable photo-catalytic performance through sunlight and antimicrobial activity. Journal of Hazardous Materials, 2020, 389: 122079. http://dx.doi.org/10.1016/j.jhazmat.2020.122079
[51] Y. Orooji, M.H. Irani-Nezhad, R. Hassandoost, et al. Cerium doped magnetite nanoparticles for highly sensitive detection of metronidazole via chemiluminescence assay. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2020, 234: 118272. http://dx.doi.org/10.1016/j.saa.2020.118272
[52] Y. Orooji, S. Mortazavi-Derazkola, S.M. Ghoreishi, et al.. Mesopourous Fe3O4@SiO2-hydroxyapatite nanocomposite: Green sonochemical synthesis using strawberry fruit extract as a capping agent, characterization and their application in sulfasalazine delivery and cytotoxicity. Journal of Hazardous Materials, 2020, 400: 123140. http://dx.doi.org/10.1016/j.jhazmat.2020.123140
[53] M. Ghasemi, A. Khataee, P. Gholami, et al. In-situ electro-generation and activation of hydrogen peroxide using a CuFeNLDH-CNTs modified graphite cathode for degradation of cefazolin. Journal of Environmental Management, 2020, 267: 110629. http://dx.doi.org/10.1016/j.jenvman.2020.110629
Copyright© Nakisa Ghamari, Shokoufeh Heydaripour, Zahra Karimi, Rozita Farhadi and Nazanin Ghamari. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.