Research Article
Design and Development of Valsartan-Loaded Solid Lipid Nanoparticles for the Enhanced Protection from Diabetic Neuropathy
Arun Radhakrishnan, Senthil Venkatachalam*, Bhavani Paruchuri
Department of Pharmaceutics, JSS College of Pharmacy, Ooty, JSS Academy of Higher Education and Research, India.
* Corresponding author. E-mail: senthil.v@jssuni.edu.in
Received: Jan. 20, 2019; Accepted: Sep. 18, 2019; Published: Sep. 20, 2019
Citation: Arun Radhakrishnan, Senthil Venkatachalam, and Bhavani Paruchuri, Design and Development of Valsartan Loaded Solid Lipid Nanoparticles for the Enhanced Protection from Diabetic Neuropathy. Nano Biomed. Eng., 2019, 11(3): 306-312.
DOI: 10.5101/nbe.v11i3. p306-312.
Abstract
Valsartan has shown to be effective against diabetic neuropathy. In this study we designed, developed and characterized valsartan loaded solid lipid nanoparticles to enhance the protection against diabetic neuropathy. Nerve function parameters and behavioural studies were performed in male mice. Valsartan-loaded solid lipid nanoparticles (VSLNs) were formulated using palmitic acid as lipid and poloxamer188 as surfactant by solvent diffusion method. VSLNs were characterized for particle size and zeta potential analysis, morphology, drug entrapment efficiency and drug loading. In vitro drug release studies were performed in phosphate buffer of pH 7.4 by using dialysis bag method. In vivo studies such as motor nerve conduction velocity, thermal nociception and mechanical nociception studies were performed with both VSLNs and pure drug. The optimized batch was the one with 1 : 2 ratio of drug and lipid, and 1% surfactant was found to have the particle size of 457 nm, zeta potential of -16 mV, entrapment efficiency of 78.9±0.05%, drug loading of 26.22±0.05% and in vitro drug release of 84.84±0.07% over a period of 24 h. Based on these results, we concluded that VSLNs showed better protection from diabetic neuropathy.
Keywords: Valsartan; Palmitic acid; Poloxamer188; Solvent diffusion method; Diabetic neuropathy
Introduction
Diabetic neuropathology (DN) is one among the common complications of polygenic disease. Inflammation is one among the main reasons behind numerous deficits seen in DN. Neuropathic pain could be a common and severely disabling state touching a lot of folks worldwide. It is characterised by the sensory abnormalities like dysesthesia, hyperalgesia and allodynia [1, 2]. Peripheral neuropathic pain is determined in patients with cancer, AIDS, long standing polygenic disease, lumbar disc syndrome, herpes infection, traumatic neural structure injury, degenerative disorder and stroke [3]. Moreover, post-thoracotomy, post-herniorrhaphy, post-mastectomy and post-sternotomy have conjointly been related to neuropathic pain [4]. Neuropathic pain responds poorly to several classical analgesics and is difficult to manage [5]. The chronic constriction injury (CCI) model is that the most typically used animal model of nerve damage-induced allodynia/hyperalgesia and has shown to share the pathophysiology with a spread of neuropathic pain conditions within the patients [6]. The nerve is entrapped through four loose ligatures during this model, and it shares the pathophysiology of carpal tunnel syndrome in humans thanks to demurrer of the median nerve within the narrowing carpal tunnel and tarsal tunnel syndrome thanks to demurrer of the leg bone nerve. What is more, this model has conjointly been prompt to share the pathophysiology of complicated regional pain syndrome in humans [6, 7]. Renin-angiotensin system plays a very important role within the body to keep up physiological state, and angiotensin II is the main final molecule of this program that in turn produces majority of its effects through activation of Hypertensin AT1 receptors. Studies have documented the presence of Hypertensin AT1 receptors on inflammatory cells [8, 9] and the important role of activation of protease Hypertensin system in several processes of inflammation as well as accumulation of neutrophils; differentiation of nerve fibre cells and production of inflammatory chemokines [10, 11]. Hypertensin AT1 receptor blockers-induced inhibition of animal tissue inflammation might in all probability be chargeable for their helpful effects in migraine [12, 13]. It has conjointly been reportable that recurrent administration of Hypertensin AT1 receptor blockers and Hypertensin changing protein inhibitors exhibit anti-nociceptive actions in mice [14]. Valsartan could be a nonpeptide, orally active and specific angiotensin II antagonist working on the AT-I receptor subtype. It is categorized as angiotensin II receptor blocker [15]. Valsartan is poorly soluble, and the liquid solubility is reportable to 1 mg/mL. Valsartan gets quickly absorbed following oral administration, with a bioavailability of 10-35% as a result of intensive internal organ first-pass metabolism. Peak plasma concentrations of valsartan accomplish at 2-4 h when administered orally dose [16]. A speedy onset of action is likely to supply quick relief within the treatment of heart condition and pain. Therefore, it is necessary to reinforce the liquid solubility and dissolution rate of valsartan to get quicker onset of action, minimize the absorption and improve its overall oral bioavailability bypassing internal organ first-pass metabolism. According to the Biopharmaceutical Classification Scheme, valsartan is taken into account as a class-II compound having less water solubility and high permeability [17, 18]. Solid lipid nanoparticles (SLNs) are a straightforward mixture drug delivery system. They are sub-micron sized (50-1000 nm) carriers created of biocompatible and perishable materials and capable of incorporating deliquescent further as hydrophobic drugs [19]. SLNs exhibit numerous benefits like enhancement of oral bioavailability, sensible physical stability, sensible tolerability, drug targeting, improved therapeutic result, protection of labile medication, possession of solid matrix controlled unleash properties, lower toxicity, potential sterilization and the simplest production scalability [20]. In this study, valsartan-loaded solid lipid nanoparticles (VSLNs) were ready to strengthen the protection from diabetic pathology. It is hypothesized that SLNs will avoid initial pass internal organ metabolism and improve the liquid solubility and dissolution rate of valsartan.
Experimental
Drugs and chemicals
Valsartan was provided by Sigma Aldrich, Mumbai. Palmitic acid and poloxamer 188 were purchased from SD Fine Chemicals, Mumbai. Streptozotocin was purchased from Sigma Aldrich, Mumbai.
Valsartan SLNs (VSLNs) formulation
Preparation of VSLNs
Valsartan loaded solid lipid nanoparticles were prepared by solvent diffusion method [21]. Lipid (plametic acid) of different concentrations was mixed with drug and dissolved in organic solvent above the melting point of lipid. Simultaneously, surfactant span 80 (0.5% w/v) and polaxomer 188 (1% w/v) solution of different concentrations was also prepared. Organic phase was added to the aqueous phase by continuous stirring. This internal phase was added to the external aqueous phase under continuous stirring using triple blade stirrer for a period of time, and the resultant nanoformulation was centrifuged and kept in refrigerator.
Characterization of VSLNs
Particle size and zeta potential analysis
The nanoparticles were characterized in terms of size (expressed as average diameter) and zeta potential using a Zeta Sizer Nano Series ® (Malvern Instruments, UK) equipped with a 4 mW He-Ne laser at a wavelength of 633 nm. Dynamic laser scattering (DLS) at a scattering angle of 173 ° was used to measure the hydrodynamic diameter (d in nm), and laser Doppler anemometry (LDA) was used to determine zeta potential in milli volts (mV) of the VSLNs. Each measurement was performed in triplicate and the data were expressed as mean ± standard deviation.
Morphology
The surface morphology of nanoparticles was determined by using scanning electron microscopy. Samples were diluted with ultra-purified water to obtain a suitable concentration. Then the samples were spread on a sample holder and dried using vacuum. They were subsequently coated with gold (JFC 1200 fine coater, Japan) and examined by a scanning electron microscope (SEM), scanned by the refractive index at 30 s run time.
Drug loading and entrapment efficiency [22]
Valsartan SLNs dispersion was centrifuged at 9000 rpm for 30 min, and the supernatant liquid was removed without disturbing the settled drug pellets. Free drug content was estimated at 249 nm using ultraviolet-visible spectrophotometer. The drug loading and entrapment efficiency were calculated by the following equations.
![]()
![]()
In vitro drug release
Dialysis method was used to determine the in vitro drug release. SLNs equivalent to 40 mg were suspended in dissolution media (phosphate buffer 7.4) and put in dialysis bags (MWCO 12000, HiMedia). The dialysis bags were placed in 100 mL of dissolution medium and stirred under magnetic stirring at 37 oC. Aliquots of the dissolution medium were withdrawn at each time interval, and the same volume of fresh dissolution medium was added to maintain a constant volume. Samples withdrawn at different time intervals were analyzed for valsartan content spectrophotometrically against solvent blank [23].
In vivo studies
Induction of diabetes and experimental design [24]
Male mice (25–27 g) were maintained at a constant temperature (23±1 °C) on a 12 h light/dark cycle with free access to food and water. Diabetes was induced by a single intraperitoneal injection of 55 mg/kg streptozotocin (STZ), in 0.1 M sodium-citrate buffer (pH 4.4). Control mice received an equivalent amount of the sodium-citrate buffer. Blood samples were collected from tail vein 48 h after STZ administration. The mice with blood glucose of more than 250 mg/dl were considered as diabetic and were further considered for the study. The experimental groups comprised of non-diabetic control mice, diabetic control mice and diabetic mice treated with (i) naïve valsartan suspended in 0.5% carboxymethyl cellulose (CMC), and (ii) valsartan SLN formulation. The treatment was started after 6 weeks of diabetes induction and was continued daily for a period of 4 weeks. The functional and behavioral experiments were performed 24 h after administration of the last dose [25]. The CPCSEA number for the study was JSSCP/IAEC/M.Pharm/
pharmaceutics/06/15-16.
Nerve function parameter
Motor nerve conduction velocity (MNCV)
MNCV was measured using Lab Chart 7 (Power Lab 4sp, Ad Instruments, Australia). Animals were anesthetized by using diethyl ether. Sciatic nerve was stimulated with 3 volt, proximally at sciatic notch and distally at ankle using bipolar needle (26½ gauge) electrodes. Receiving surface electrodes were placed on the foot muscle. The latencies of the compound muscle action potentials were recorded via bipolar electrodes from the first interosseous muscle of the hind paw and measured from the stimulus artifact to the onset of the negative M-wave deflection. MNCV was calculated by dividing the distance between the stimulating and recording electrode by difference in distal and proximal latency. MNCV was expressed in m/s [26].
Behavioral Studies
Thermal hyperalgesia
Thermal hyperalgesia of the tail to both hot (45 oC) and cold (10 oC) immersion was studied. Rats were acclimatised three days prior to the experiment. The tail flick response latency or any signs of struggle was observed as the endpoint response. The cut-off time was kept as 15 s in both tests. Three consecutive readings were taken at an interval of 10 min [27].
Mechanical hyperalgesia
Sensitivity to noxious mechanical stimuli was determined by quantifying the withdrawal threshold of the hind paw in response to mechanical stimulation using a Randall Selitto callipers as described previously [28].
Results and Discussion
Particle size and zeta potential analysis
The mean particle size of VSLNs was found to be 457.1 nm and the zeta potential was found to be -16 mV. Polydispersity index (PDI) values were found to be 0.320, which showed that the system had a relatively narrow size distribution (Fig. 1). The zeta potential of the prepared nanoparticles was the prominent factor to ensure stability. According to Nanotechnology Characterization Laboratory PDI, the broadness of size distribution was measured (Fig. 2). As a rule of thumb, PDI values smaller than about 0.30 were considered monodisperse and stable [29].
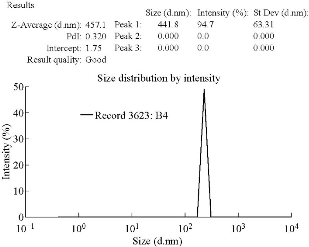
Fig. 1 Size and PDI report for VSLNs.
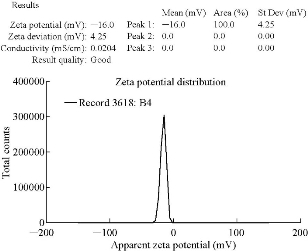
Fig. 2 Zeta potential report for VSLNs.
Morphology
Scanning electron microscopy was used to verify the uniformity of particle shape and size (Fig. 3). The SEM microphotographs of valsartan loaded solid lipid nanoparticles revealed that the nanoparticles were spherical in shape having smooth surface [30].
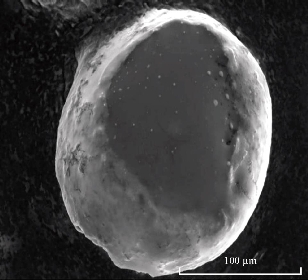
Fig. 3 Scanning electron micrograph of valsartan loaded solid lipid nanoparticles.
Drug loading and entrapment efficiency
Percentage of entrapment efficiency (EE) for optimized SLNs formulation was found to be 78.9±0.05%. High percentage of EE could be attributed to the lipophilic nature of drug, as it had higher affinity for the lipid matrix. Drug loading for optimized SLNs formulation was found to be 26.22±0.05% [31].
In vitro drug release studies
In vitro release studies of valsartan loaded solid lipid nanoparticles were carried out in pH 7.4 phosphate buffer; the release profile of optimized batch B4 was observed up to 24 h. The release profile of this batch was compared with the pure drug (Fig. 4). Among all, nanoformulation was found to exhibit satisfactory sustained release profile, which it indicated sustained release of drug from the SLN formulation. The experiment was repeated for 3 times (n = 3), and the average value was taken into consideration.
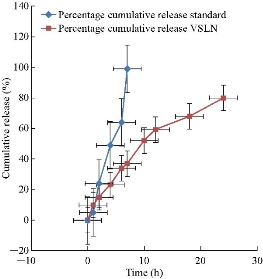
Fig. 4 Drug release profiles of B4 formulation and pure drug.
In vivo studies on effect of valsartan on body weights of mice
Baseline values of body weights were measured before starting experiments, and the average body weight was found to be 23.2±0.8 g. Body weight changes of all groups are shown in Fig. 518. Body weights of the STZ induced group, pure valsartan and valsartan SLN formulation groups were significantly (p < 0.001) lower than those of the normal group on days 7, 14, 21 and 28. However, there was no significant difference in body weights between the pure drug and the formulation treated group [32].
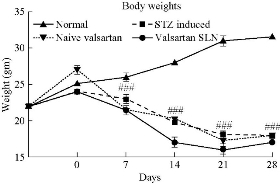
Fig. 5 Effect of VSLNs on body weight changes.
Effect of VSLNs on motor nerve conduction velocity (MNCV)
Motor nerve conduction velocity was measured 24 h after the last dose of STZ induced diabetic neuropathy. It resulted in significantly reduced nerve conduction velocity in sciatic nerve (Table 1). Treatment was given with naïve valsartan and valsartan SLN. Valsartan SLN treatment significantly reversed the altered nerve conduction velocity in sciatic nerve (Fig. 6).
Table 1 Effect of VSLNs on altered NCV in STZ induced mice
|
Treatment group |
MNCV (m/s) |
|
Normal |
37.4 ± 1.6 |
|
STZ |
18 ± 1.9 ### |
|
Naïve valsartan |
34 ± 2.3 ** |
|
Valsartan SLN |
36 ± 1.7 ***, @@ |
Note: Data is expressed as mean ± SEM.
# p < 0.05 vs. normal.
** p < 0.01 and *** p < 0.001 vs. STZ induced group.
@@ p < 0.01 vs. naïve valsartan treated group.
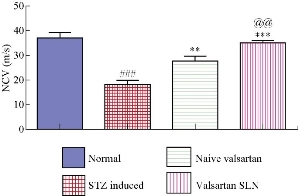
Fig. 6 Effect of VSLNs on motor nerve conduction velocity in STZ treated mice.
Effect of VSLNs on thermal nociceptive threshold
STZ administration significantly lowered the tail-flick latency (both hot and cold) compared with the normal group on days 14, 21 and 28 (p < 0.05). Naïve valsartan and valsartan SLN groups showed a statistically significant increase of tail withdrawal thresholds compared with the STZ group on days 14, 21 and 28 (p < 0.05). Tail flick latencies are shown in Fig. 7(a) and (b).
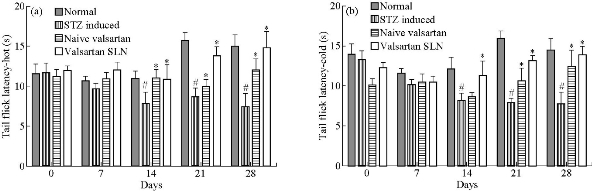
Fig. 7 Effect of VSLNs on (a) hot and (b) cold tail-flick latency on days 0, 7, 14, 21 and 28 of STZ induced group; naïve valsartan and valsartan SLN treated groups respectively.
Effect of VSLNs on mechanical nociceptive threshold
Mechanical nociceptive threshold changes in the paw pressure test are shown in Fig. 8. No significant difference was observed on day 0. STZ significantly lowered paw withdrawal thresholds compared to the normal group on days 7, 14, 21 and 28 (p < 0.05 on days 21 and 28). There was statistically significant improvement of mechanical nociceptive threshold observed with the treatment of pure valsartn and the valsartan SLN formulation.
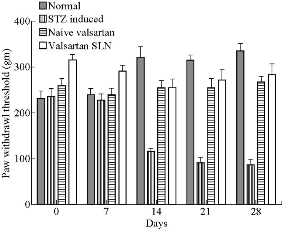
Fig. 8 Effect of VSLNs on mechanical hyperalgesia (Randall Sellito test) on days 0, 7, 14, 21 and 28.
Conclusions
Diabetic neuropathy (DN) is associated with various sensorimotor, functional and biochemical alterations. Sensory alteration was evidenced by thermal and mechanical hyperalgesia. Valsartan treatment reversed these sensorimotor deficits in experimental DN and more significantly with solid lipid nanoparticles (SLNs) formulation. The efficacy of valsartan is limited because of its low aqueous solubility and poor bioavailability due to the first-pass metabolism. Different studies have been carried out to improve the aqueous solubility. Valsartan-loaded solid lipid nanoparticles (VSLNs) could improve the solvability of valsartan and reduce the first-pass metabolism by enhancing the protection from diabetic neuropathy, to reduce neuro-inflammation by achieving sustained release of valsartan using solid lipid nanoparticles. Batch prepared with 1 : 2 ratio of drug : lipid, with 1% concentration of surfactant (B4) showed the minimum particle size as of 457.1 nm, zeta potential as of -16 mV and PDI 0.032, which was identified as an ideal batch. In vitro drug release studies were performed for VSLNs batch B4 and pure valsartan. It was observed that the VSLNs formulation showed sustained drug release over a period of 24 h, while the pure drug exhibited a drug release for 5 h. It is concluded that the solvent diffusion method is suitable for producing valsartan-loaded solid lipid nanoparticles. Lipophilic drugs like valsartan can be easily incorporated into the lipid matrix using this method.
The formulated VLSNs showed better protection from diabetic neuropathy compared to pure drug. Neuroprotection action was observed as significantly higher with the VSLNs formulation when compared with the pure drug. VSLNs exhibited a nerve conduction velocity of 36 ± 1.7 m/s, while the values for the naïve valsartan, STZ and normal group were 34 ± 2.3 m/s, 18 ± 1.9 m/s and 36.4 ± 1.6 m/s, respectively. The VSLNs reversed the altered nerve conduction velocity in sciatic nerve as compared with the other groups. The tail flick latency - hot and tail flick latency - cold were minimal for the VSLNs treated group. From the data it was revealed that the prepared VSLNs exhibited a significantly greater in vivo characteristics as compared to the reference, which represents an enhanced neuro-protective activity of the formulation.
References
Copyright© Arun Radhakrishnan, Senthil Venkatachalam, and Bhavani Paruchuri. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
|
Nano Biomedicine and Engineering. Copyright © Shanghai Jiao Tong University Press |
 |